
Abstract
Conjugated linoleic acid (CLA) is thought to have anti-proliferative and anti-inflammatory properties, but its effect on cancer cachexia is unknown. Two effects were here investigated: that of CLA on inflammatory mediator production in human lung cancer cells, and that of reduced mediators on the myogenic differentiation of murine muscle C2C12 cells. The latter cells were grown in medium conditioned by human lung cancer A427 cells, with or without CLA, to mimic only the effect of molecules released from the tumor “in vivo”, excluding the effect of host-produced cachectic factors. The results obtained show that CLA was found to reduce the production of tumor necrosis factor-α, interleukin (IL)-1β and prostaglandin E2 (PGE2), but had no effect on IL-6 production. The mechanisms underlying the effect of CLA on cytokine or PGE2 release in A427 cells are probably mediated by activation of peroxisome proliferator-activated receptor (PPAR)α, which increased at 24 h CLA treatment. In turn, the reduced content of inflammatory mediators in medium conditioned by A427 cells, in the presence of CLA, allowed muscle cells to proliferate, again by inducing PPAR. The involvement of PPARα was demonstrated by treatment with the antagonist MK-886. The findings demonstrate the anti-inflammatory and myogenic action of CLA and point to its possible application as a novel dietary supplement and therapeutic agent in inflammatory disease states, such as cachexia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the commonest manifestations of advanced malignant disease is the development of cancer cachexia, which is responsible for about 20 % of cancer-related deaths. Associated abnormalities include anorexia, weight loss, muscle loss and atrophy, anemia, and alterations in carbohydrate, lipid and protein metabolism [1–3]. Muscle wasting is the most important feature of cancer cachexia, and the principal cause of morbidity and mortality in cancer patients [4]. Skeletal muscle is a highly adaptable tissue, which responds to physiological challenges by changing size and metabolism, undergoing changes in growth and development under the strict control of myogenic regulatory factors [5].
Although cachectic factor(s) have long been sought, and considerable scientific effort and economic resources have been devoted to their investigation, the picture is still fragmentary. Experimental research and clinical studies have demonstrated that cytokines are involved in inducing cachexia [6, 7], although the results require careful interpretation. Episodic administration of tumor necrosis factor (TNF) has proved unsuccessful at inducing cachexia in experimental animals; repetitive TNF administration initially induces a cachectic effect, but tolerance to the cytokine soon develops and food intake and body weight return to normal. Escalating doses of TNF have been shown necessary to maintain the cachectic effect [3].
Treatment with anti-mouse interleukin (IL)-6 antibody, or with nonpeptide IL-6 receptor antagonist, successfully reversed the key parameters of cachexia in colon-adenocarcinoma-bearing mice [8, 9]. Conversely, studies on incubated rat skeletal muscle have clearly shown that IL-6 has no direct effect on muscle proteolysis [10]. IL-1β production in human and animal cancer cell lines is well documented, and patients with IL-1β-producing tumors generally have a poor prognosis [11]. This cytokine is also reported to be responsible for enhanced protein degradation in rats, in which repeated administration of IL-1β induced anorexia and weight loss [11].
Since cancer cachexia is regarded as a chronic inflammatory condition, in which pharmacological and nutritional interventions aimed at increasing muscle mass and weight are of limited benefit [12], the current strategies for managing this condition focus on the use of agents with anti-inflammatory and nutritional properties. Conjugated linoleic acid (CLA) comprises a group of positional and geometric isomers of linoleic acid, found naturally in meat and dairy products from ruminant animals. It has shown promise both in reducing inflammation and as an anti-carcinogenic agent [13, 14]. CLA is formed in the rumen during a process of bacterial biohydrogenation of linoleic acid. Although 28 different isomers of CLA exist, the c9,t11 form accounts for more than 90 % of the CLA intake in the human diet [13]. The health benefits deriving from CLA have been attributed to the two main isomer forms, c9,t11 and t10,c12; these are the predominant biologically active forms and exhibit numerous physiological properties [15, 16]. The health-related effects of CLA have been confirmed in numerous animal and human-intervention studies; they include anti-carcinogenic, anti-atherogenic, anti-diabetogenic, and immunomodulatory actions [17–19]. In vitro studies have shown CLA to inhibit proliferation of several human tumor cell lines, in a dose- and time-dependent manner [17, 20, 21].
It has been suggested that the anti-carcinogenic properties of CLA stem from its anti-inflammatory properties, as the acid negatively regulates the expression of pro-inflammatory genes such as TNF-α, IL-1β, and IL-6 [22]. Due to its anti-proliferative and potentially anti-inflammatory properties, CLA might be a novel dietary supplement for patients suffering from muscle wasting in chronic inflammatory-related disease, particularly cancer cachexia.
CLA is not universally considered to be of use as a supplement in cachexia patients [23]. The addition of c9,t11-CLA-rich oil to the diet did not ameliorate wasting in mice with cancer cachexia [24]. However, other studies have shown that CLA can preserve muscle mass in tumor-bearing cachectic animals, decreasing skeletal muscle wasting and reducing the catabolic effects of TNF-α, and have suggested that it may have overall protective properties in cachexia [25, 26]. In particular, one study reported CLA to possess specific tumor-related effects on myogenic and inflammatory gene expression, in an in vitro human co-culture model comprising a pancreatic tumor cell line and human primary muscle cells [27].
The present study investigated the effects of CLA on the proliferation and production of inflammatory mediators in human lung cancer A427 cells. It also examined the effects of the secretion products of cancer cells, both treated and untreated with CLA, on the myogenic differentiation of murine muscle C2C12 cells.
Materials and Methods
A427 Cell Culture Conditions
Human lung A427 cells (ATCC, MD, USA) were cultured (20,000 cells/cm2) in DMEM/F12 medium supplemented with 2 mM glutamine, 1 % antibiotic/antimycotic solution and 10 % FBS (medium A).
Treatment of A427 Cells with CLA
CLA was prepared in horse serum (HS) at a concentration of 5 mM. A mixture of the two principal CLA isomers, c9,t11 and t10,c12 (Sigma, MO, USA) was used. Twenty-four hours after cell seeding, medium A was removed and replaced with DMEM/F12 medium supplemented with 2 mM glutamine, 1 % antibiotic/antimycotic solution, 2 % HS (medium B), and CLA (final concentration 5 or 50 μM). In control cells, a quantity of HS equivalent to the highest dose administered with CLA was added to the cell culture, in addition to the 2 % HS present in the medium B.
Twenty-four hours after CLA or HS addition, the culture media were collected, pooled, and centrifuged at 2,800g for 10 min (centrifuge J6B Beckman Culture, CA, USA) at room temperature. The resulting medium was used as conditioned medium in which to culture C2C12 cells; it was also used to determine pro-cachectic factor production and lactate dehydrogenase (LDH) release. Cells were washed with PBS, trypsinized and centrifuged at 900g for 10 min (centrifuge J6B Beckman Culture, CA, USA) for the assays listed below.
Treatment of A427 Cells with CLA and MK886
A427 cells were divided into four groups: (1) control cells; (2) CLA group: cells treated with 5 μM CLA; (3) MK886 group: cells treated with 5 μM MK886 dissolved in DMSO; (4) CLA plus MK886 group: cells treated with both substances. After 24 h, in all groups cells were trypsinized, centrifuged at 900g (centrifuge J6B Beckman Culture, CA, USA) for 10 min, and cell proliferation was evaluated; the culture medium was used to evaluate the production of TNF-α and prostaglandin E2 (PGE2), and to culture C2C12 cells.
C2C12 Cell Culture Conditions
Murine muscle C2C12 cells (ATCC, MD, USA) were seeded at 6,000 cells/cm2 in medium A and maintained for 4 days. After this time, to induce differentiation into myotubes, medium A was removed and replaced with medium B, in which the C2C12 cells were maintained for a further 3 days. Cells were then subdivided into five groups: group I (Tdiff): cells grown in medium B; group II (control): cells grown in medium conditioned by A427 cells; group III (5 μM CLA): cells grown in medium conditioned by A427 cells treated with 5 μM CLA; group IV (MK886): cells grown in medium conditioned by A427 cells treated with 5 μM MK886; group V (MK886 + CLA): cells grown in medium conditioned by A427 cells treated with 5 μM CLA plus 5 μM MK886.
Cells and culture media of these groups were analyzed after 1 or after 4 days. After medium removal, cells were maintained frozen until use. The culture medium was centrifuged at 2,800g (centrifuge J6B Beckman Culture, CA, USA) for 10 min at 4 °C and used to determine LDH release.
Cell Growth and Viability
The number of A427 cells in the monolayer and that in the culture medium were evaluated using a Burker chamber, and expressed as cells/cm2. For both A427 and C2C12 cells, viability was evaluated by determining LDH release, as described elsewhere [28].
Immunofluorescence Staining
To evaluate production of myosin and myofibrillar structures, C2C12 cells were washed with PBS, fixed with acetone and methanol (1:1) at 20 °C for 20 min and kept at 4 °C until use. Cells were then permeabilized with 0.1 % Triton X-100 in PBS and incubated with the monoclonal anti-skeletal (fast) myosin antibody (1:400 in PBS-Triton 0.1 %) (Sigma, MO, USA) overnight at 4 °C. After washing in PBS-Triton 0.1 % and then in PBS alone, cells were incubated with the appropriate Cy3-conjugated secondary antibody (1:1000) (GE Healthcare, UK) for 1 h at room temperature.
To visualize nuclei, cells were stained with Hoechst dye for 20 min. After incubation, cells were thoroughly washed with bidistilled H2O and mounted with glycerol/PBS (1:1). Fluorescence-labeled cells were viewed using a fluorescence reverted microscope, and photographed using a ×32 lens (Zeiss, Germany). For image processing, Zeiss AxioVision software was used.
Western Blot Analysis
A427 cells were suspended in a lysis buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1 mM PMSF, 0.015 mg/ml leupeptin, 0.1 % NP-40 substitute, and 1 mM Na-orthovanadate. Suspensions were kept on ice for 30 min. C2C12 cells were rubberized and suspended in a RIPA buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 % NP-40 substitute, 0.5 % Na-deoxycholate, 0.1 % SDS, 1 mM PMSF, 1 mM Na-orthovanadate and 0.015 mg/ml leupeptin. Suspensions of both cell types were kept on ice for 10 min and sonicated. Forty microgram of cell proteins were separated by SDS–polyacrylamide gel electrophoresis, then electrotransferred to a polyvinylidene difluoride (PVDF) membrane, which was then blocked overnight with TBS containing 5 % non-fat dry milk. The membrane was then rinsed and treated with polyclonal anti-PPARα, anti-PPARβ, or anti-PPARγ antibodies (Santa-Cruz Biotechnology Inc., CA, USA), and monoclonal anti-NF-kBp65, anti-cyclooxygenase (COX)-2, anti-skeletal (fast) myosin, anti-β-actin or anti-α-tubulin antibodies (Sigma, MO, USA). Protein bands were visualized using a chemiluminescent detection system (Immun-StarTM HRP Luminol/Enhancer, Bio-Rad Laboratories Inc., CA, USA).
Real-Time PCR
A427 cells were processed to determine PPAR expression by real-time PCR. Total RNA was extracted from the cells using the NucleoSpin RNA II Kit (Macherey–Nagel GmbH & Co. KG, Germany). Real-time PCR was run with single-stranded cDNA prepared from total RNA (1 μg) using a High-Capacity cDNA Archive kit (Applied Bio Systems, CA, USA). The forward (FW) and reverse (RV) primers were designed using the Beacon Designer® software package (Bio-Rad Laboratories Inc., CA, USA). Twenty-five microliters of a PCR mixture, containing cDNA template equivalent 40 ng of total RNA, 5 pmol each of forward and reverse primers, and 2× IQ SYBR Green SuperMix (Bio-Rad Laboratories Inc., CA, USA), were amplified in an iCycler PCR instrument (Bio-Rad Laboratories Inc., CA, USA) with an initial 10 min melt at 95 °C, followed by 35–40 cycles at 95 °C for 40 s, annealing temperature for each primer set for 40 s, and 72 °C for 40 s. Each sample was tested in duplicate, and threshold cycle (Ct) values were averaged. The variation in expression was defined as the relative expression in cells treated with CLA versus that in control cells, calculated as 2−∆∆Ct, where ∆Ct = CtCLA – CtGAPDH and ∆∆Ct = ∆CtCLA – ∆Ctcontrol.
Chemical Analysis
TNF-α, IL-1β and IL-6 were evaluated by the Enzyme Linked Immuno Sorbent Assay (Bender Medsystems, Austria), while PGE2 was evaluated with an Enzyme Immunoassay Kit (Oxford Biomedical Research, MI, USA).
Protein Determination
Protein concentrations in A427 and C2C12 cell lysates were measured using the Protein Assay Kit 2, following the manufacturer’s instructions (Bio-Rad Laboratories Inc., CA, USA).
Statistical Analysis
All data are expressed as mean ± SD differences between group means were assessed by analysis of variance followed by a post hoc Newman-Keuls test or by the Student’s t test.
Results
Effect of CLA on Human Lung Cancer A427 Cells
After 24 h, only the highest concentration of CLA (50 μM) significantly reduced the number of cells, whereas the lowest concentration (5 μM) left cell numbers similar to control values (Fig. 1). Neither concentration of CLA induced death, by necrosis or by apoptosis, as demonstrated by LDH release in the medium and by cytofluorimetric analysis (data not shown). For subsequent determinations, only the 5 μM CLA concentration was used, in order to avoid as far as possible any interference from decreased cell proliferation.

Numbers of human lung cancer A427 cells, untreated or treated with MK886, CLA or CLA + MK886 for 24 h. Data are mean ± SD from four experiments. Means with different letters are significantly different from one another (p < 0.05) as determined by analysis of variance followed by post hoc Newman–Keuls analysis. C control cells, CLA5 cells treated with 5 μM CLA, CLA50 cells treated with 50 μM CLA, MK5 cells treated with 5 μM MK886, CLA5 + MK5 cells treated with 5 μM CLA + 5 μM MK886
Evaluation of cytokines in the culture medium (Fig. 2) shows that CLA caused a significant decrease of TNF-α and IL-1β versus control cells at 24 h, with no significant decrease of IL-6. Figure 2 also shows that the release of PGE2 in the culture medium was lower in cells treated with CLA than in control cells.

Concentrations of TNF-α (a), IL-1β (b), IL-6 (c) and PGE2 (d) in medium of human lung cancer A427 cells, untreated or treated with CLA for 24 h. Data are mean ± SD from four experiments. Student’s t test: *p < 0.05, control cells versus cells treated with 5 μM CLA. C control cells, CLA5 cells treated with 5 μM CLA
Effect of Medium Conditioned by A427 Cells on Murine Muscle C2C12 Cells
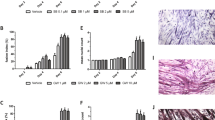
Medium conditioned for 24 h by A427 cells, untreated or treated with CLA, did not affect the viability of C2C12 cells, as evidenced by LDH determination in the culture medium (data not shown). Figure 3 shows the differentiation degree of C2C12 cells, grown for 1 or for 4 days in conditioned medium; number and size of myotubes were evidenced by immunofluorescence staining. At both 1 and 4 days, cells grown in medium conditioned by A427 cells without CLA showed little or no differentiation, compared to cells induced to differentiate in the appropriate medium (Tdiff). Conversely, cells grown for 4 days in medium conditioned by A427 cells treated with 5 μM CLA showed differentiation increased in a time-dependent manner, reaching almost the same degree of differentiation as cells grown in Tdiff. These results were confirmed by evaluating myosin protein expression, which significantly increased in C2C12 cells grown for 4 days, in medium conditioned by A427 cells with 5 μM CLA, compared to control cells (Fig. 3).

Myosin content evidenced by immunofluorescence staining and by Western blot analysis in C2C12 murine myoblasts after 1 or 4 days of differentiation. Differentiation was in medium conditioned by human lung cancer A427 cells, untreated or treated with CLA for 24 h. For Western blot analysis, 40 μg of cell proteins were applied for each lane. Tdiff cells grown in medium b; C cells grown in conditioned medium removed from A427 cells untreated with CLA; CLA5 cells grown in conditioned medium removed from A427 cells treated with 5 μM CLA
PPAR Involvement
CLA is a PPAR ligand; these receptors were thus examined in A427 cells, to investigate their possible involvement in the CLA-induced decrease of cytokine and PGE2 production. Figure 4 shows the PPAR trend, evaluated as mRNA (Fig. 4a) and protein expression (Fig. 4b). Both measures of PPARα significantly increased versus control cells, 24 h after treating A427 cells with CLA; conversely, PPARγ was unchanged, and PPARβ showed a small but not significant increase. As Fig. 4c shows, the protein expression of NF-kB and COX-2 decreased in CLA-treated cells versus controls cells after 24 h treatment.

PPAR, COX-2 and NF-kBp65 expression in human lung cancer A427 cells, untreated or treated with CLA for 24 h. PPARs were analyzed by real-time PCR (a) and Western blot (b), COX-2 and NF-kBp65 were analysed by Western blot (c). Data are mean ± SD from three experiments. For real-time PCR (1 μg of cDNA), the values of cells treated with CLA are referred to control value, taken as 1 (white bar). For Western blot (40 μg of cell proteins for each lane), the densitometry value given for each protein is normalized to the corresponding β-actin value and expressed as a percentage, setting the control value (C) arbitrarily at 100. Student’s t test: *p < 0.05, control cells versus cells treated with 5 μM CLA. C control cells, CLA5 cells treated with 5 μM CLA, C+ positive control
Since PPARs are involved in reducing the inflammatory process, their influence in regulating myosin production in C2C12 cells was also examined. Figure 5 shows the PPAR trend evaluated as mRNA (Fig. 5a, b) and as protein (Fig. 5c): PPARα increased in C2C12 cells grown for 1 day in the culture medium of A427 cells treated with CLA, whereas no variation was found at 4 days. PPARγ remained unchanged. As regards PPARβ, a small but not significant increase occurred after 1 day, and a significant increase after 4 days.

PPAR expression evidenced by real-time PCR (a, b) and Western blot (c) in murine muscle C2C12 cells after 1 and 4 days of differentiation. Differentiation was in medium conditioned by human lung cancer A427 cells, untreated or treated with CLA for 24 h. Data are mean ± SD from three experiments. For real-time PCR (1 μg of cDNA), the values of cells treated with CLA are referred to the control value, taken as 1 (white bar). For Western blot (40 μg of cell proteins for each lane), the densitometry value given for each protein is normalized to the corresponding β-actin value and expressed as a percentage, setting the control value (C) arbitrarily at 100. For each PPAR, means with different letters are significantly different from one another (p < 0.05) as determined by analysis of variance followed by post hoc Newman–Keuls test. C cells grown in conditioned medium removed from A427 cells untreated with CLA, CLA5 cells grown in conditioned medium removed from A427 cells treated with 5 μM CLA
To investigate the relation between PPARα and cytokine and PGE2 production in A427 cells, a concentration of PPARα-antagonist MK886 that does not affect cell proliferation was administered to the cells (Fig. 1). The presence of MK886 prevented the reduction in TNF-α and PGE2 production in A427 cells (Fig. 6), and the increase of number and size of myotubes at both 1 and 4 days in C2C12 cells, as evidenced by immunofluorescence staining (Fig. 7).

Concentrations of TNF-α (a) and PGE2 (b) in medium of human lung cancer A427 cells, untreated or treated with CLA and MK886 for 24 h. Data are means ± SD from three experiments. For each panel, means with different letters are significantly different from one another (p < 0.05) as determined by analysis of variance followed by post hoc Newman–Keuls test. C control cells, CLA5 cells treated with 5 μM CLA, MK5 cells treated with 5 μM MK886, CLA5 + MK5, cells treated with 5 μM CLA + 5 μM MK886

Myosin content evidenced by immunofluorescence staining in murine muscle C2C12 cells after 4 days of differentiation. Differentiation was in medium conditioned by human lung cancer A427cells, untreated or treated with MK886, CLA, or CLA + MK886 for 24 h. C cells grown in conditioned medium removed from A427 cells untreated with CLA, CLA5 cells grown in conditioned medium removed from A427 cells treated with 5 μM CLA, MK5 cells grown in conditioned medium removed from A427 cells treated with 5 μM MK886, CLA5 + MK5 cells grown in conditioned medium removed from A427 cells treated with 5 μM CLA + 5 μM MK886
Discussion
The study provides some insight into anti-inflammatory and myogenic effects of the fatty acid CLA, and provides a glimpse of its possible applications as a therapeutic agent in inflammatory states underlying cachexia in cancer patients. An in vitro model in which murine muscle C2C12 cells were grown in a medium conditioned by A427 human lung cancer cells, exposed or not to CLA, was used to mimic the effect of molecules released from the tumor in vivo; this excluded any possible interference of the effects of cachexia-mediator factors produced by the host. One difficulty of in vivo research is that of differentiating between effects due to pro-cachectic mediator factors produced by the host, and those produced by cancer cells. A mixture of c9,t11 and t10,c12 CLA isomers was used, because they are the two major CLA forms present in the diet and showing biological activity, even if c9,t11 CLA constitutes up to 80 % of total CLA.
Exposing A427 cells to CLA reduced production of TNF-α, IL-1β and PGE2, the reduction of PGE2 being due to the decrease of COX-2. COX-2 is the pro-inflammatory COX isoform, and is the rate-limiting enzyme in the synthesis of prostaglandin PGE2 from arachidonic acid, one of many fatty acids in the phospholipids of cell membranes [29].
In murine muscle C2C12 cells, medium removed from A427 cells not treated with CLA did not enable differentiation to occur, as evidenced by the minimal number and size of myotubes (immunofluorescence staining) and low level of myosin expression (Western blot). Conversely, medium conditioned by the presence of CLA, which had a lower content of pro-inflammatory mediators than control medium, allowed time-dependent differentiation to occur, evidencing the importance of these factors on muscle cell modifications. In our study, CLA mixture administered to A427 cells enabled C2C12 cell differentiation, despite report that only the c9,t11 isomer stimulates C2C12 cell differentiation [30]. It appears probable that, in the case of administration of a CLA mixture, the effect of the c9,t11 isomer prevails.
There is considerable evidence from animal studies that TNF-α plays a role in muscle loss in cancer cachexia, although its role in the human condition is more questionable [31, 32]. Be that as it may, in our model the reduction of PGE2, coupled with the reduction of cytokines, regardless of cytokine type, was associated with muscle cell differentiation. Mechanisms thought to underlie the effect of CLA on the decrease of cytokines or of PGE2 in A427 cells were mediated by activation of PPARα, which increased after 24 h of CLA treatment, whereas the other PPARs did not show any statistically significant variations.
The PPAR family comprises three ligand-activated transcription factors: PPARα (NR1C1), PPARβ (NR1C2), and PPARγ (NR1C3). All three PPARs play important roles in regulating metabolic pathways, including those of lipids and glucose. They are also involved in cell differentiation, proliferation, and apoptosis pathways, and great interest has been shown in their involvement in inflammatory processes. In particular, PPARα and PPARγ inhibit the activation of inflammatory gene expression, and may negatively interfere with pro-inflammatory signaling pathways in vascular and inflammatory cells. The role played by PPARβ in regulating inflammation and immunity is only now emerging [33].
PPARα agonist-induced activation has been shown to inhibit a number of inflammatory pathways, including TNF-α, inducible nitric oxide synthase, and COX-2, and that it also inhibits infiltration of adhesion molecules and cells into the tissues [34]. In this study, inhibition of TNF-α, IL-1β and PGE2 by PPARα may have been mediated by the decrease of p65 NF-kB, a hypothesis that would agree with reports showing that increased PPARα expression may inactivate TNF-α and COX-2, by inhibiting NF-kB. PPARα translocates from the cytosol to the nucleus, where it binds with NF-kB to inhibit gene activation [35–37]. The promoter regions of TNF-α, IL-1β and COX-2 genes contain numerous transcription-factor binding sites, including AP-1 and NF-kB. Several lines of evidence suggest that PPARα exerts its anti-inflammatory effects through negative regulation of NF-kB. In human aortic smooth-muscle cells, PPARα has been shown to inhibit the NF-kB transactivation function, by directly interacting with the Rel homology domain of the p65 subunit, which mediates DNA binding and dimerization activity [38]. PPARα ligands have also been found to upregulate expression of the NF-kB inhibitor protein IkBα, in both human aortic smooth-muscle cells and microglial cells, leading to decreased DNA binding of NF-kB and gene expression [39, 40].
The involvement of PPARα in reduction of inflammatory factors induced by CLA was confirmed by using the PPARα antagonist MK886, which enabled TNF-α and PGE2 release to return to near control values in A427 cells, while in C2C12 cells it permitted the anti-miogenic effect of cytokines to occur. In our experimental model, PPARα was involved not only in the effect of CLA on A427 cells, but probably also in the effect of cytokines and PGE2 on C2C12 cell differentiation. In the latter cells, medium conditioned by A427 treated with CLA, and with a lower content of pro-inflammatory factors than the medium of untreated cells, caused an increase in PPARα, especially at 1 day, and in PPARβ at 4 days. The increased PPAR expression might be the cause of the increased myosin content and increased number and size of myotubes, that occurred in C2C12 cells grown in medium with reduced inflammatory mediator content. The increase in PPAR caused by the reduction of cytokines and PGE2 could also be explained by the inhibition of NF-kB: TNF-α induces muscle protein degradation by activating NF-kB, dependently of ROS production or otherwise [31, 41].
The physical interaction between PPAR and the p65 subunit of NF-kB interferes with the activity of both transcription factors [38, 40]. Because interaction between PPAR and NF-kB leads to modulation of the inflammatory response, a reciprocal effect of NF-kB on the PPAR transactivation function is obviously to be expected. In vitro assays have shown that translocation of NF-kB into the nucleus represses PPAR transactivation of a PPRE-driven promoter [42]. Increased PPAR expression may explain the positive effect of reduced inflammatory mediator content on increasing myosin content, number and size of myotubes.
PPARβ activation has been shown not only to increase the number of oxidative myofibres and capillaries in skeletal muscle, but also to promote myonuclear accretion [43], which is a characteristic response to exercise [44–46]. Increased myonuclear density has been found in transgenic muscle-specific over-expressing animals, as well as in wild-type mice treated for only 2 days with a PPARβ agonist. As mouse aging is associated with reduced PPARβ expression, and with decreased numbers of oxidative myofibres and myonuclei, which could be partially reversed by pharmacological PPARβ activation, a potential use of PPARβ agonists for the treatment of age-related muscle atrophy has been suggested [43].
In conclusion, CLA was here found to reduce release into the culture medium of inflammatory factors, produced by lung cancer cells, through activation of PPAR. In turn, the reduced content of these factors enabled differentiation of muscle cells, again through induction of PPAR. The study provides insight into the anti-inflammatory and myogenic actions of CLA, and points to its possible application as a therapeutic agent in inflammatory disease states, such as cachexia.
Abbreviations
- CLA:
-
Conjugated linoleic acid
- COX:
-
Cyclooxygenase
- IL:
-
Interleukin
- LDH:
-
Lactate dehydrogenase
- PGE2:
-
Prostaglandin E2
- PPAR:
-
Peroxisome proliferator-activated receptor
- TNF:
-
Tumour necrosis factor
References
Costelli P, Baccino FM (2000) Cancer cachexia: from experimental models to patient management. Curr Opin Clin Nutr Metab Care 3:177–181
Fearon K, Moses A (2002) Cancer cachexia. Int J Cardiol 85:73–81
Argiles JM, Busquets S, López-Soriano FJ (2005) The pivotal role of cytokines in muscle wasting during cancer. Int J Biochem Cell Biol 37:2036–2046
Tisdale MJ (1997) Cancer cachexia: metabolic alterations and clinical manifestations. Nutrition 13:1–7
Hawke TJ, Garry DJ (2001) Myogenic satellite cells: physiology and molecular biology. J Appl Physiol 91:534–551
Argiles JM, Busquets S, Toledo M, López-Soriano FJ (2009) The role of cytokines in cancer cachexia. Curr Opin Support Palliat Care 3:263–268
Kemik O, Sumer A, Kemik AS, Itik V, Dulger AC, Purisa S, Tuzun S (2010) The relationship among acute-phase response proteins, cytokines and hormones in cachectic patients with colon cancer. World J Surg Oncol 28:8–85
Strassmann G, Fong M, Freter CE, Windsor S, D’Alessandro F, Nordan RP (1993) Suramin interfers with interleukin-6 receptor binding in vitro and inhibits colon-26-mediated experimental cancer cachexia in vivo. J Clin Invest 92:2152–2159
Enomoto A, Rho MC, Fukami A, Hiraku O, Komiyama K, Hayashi M (2004) Suppression of cancer cachexia by 20S, 21-epoxy-resibufogenin-3-acetate-a novel nonpeptide IL-6 receptor antagonist. Biochem Biophys Res Commun 323:1096–1102
Garcıa-Martınez C, Lopez-Soriano FJ, Argiles JM (1994) Interleukin-6 does not activate protein breakdown in rat skeletal muscle. Cancer Lett 76:1–4
Zhang D, Zheng H, Zhou Y, Tang X, Yu B, Li J (2007) Association of IL-1beta gene polymorphism with cachexia from locally advanced gastric cancer. BMC Cancer 14:7–45
Tisdale MJ (2001) Cancer anorexia and cachexia. Nutrition 17:438–442
Bhattacharya A, Banu J, Rahman M, Causey J, Fernandes G (2006) Biological effects of conjugated linoleic acids in health and disease. J Nutr Biochem 17:789–810
MacDonald HB (2000) Conjugated linoleic acid and disease prevention: a review of current knowledge. J Am Coll Nutr 19:111–118
Belury M (2002) Dietary conjugated linoleic acid in health: physiological effects and mechanisms of action. Annu Rev Nutr 22:505–531
Pariza MW, Park Y, Cook ME (2001) The biologically active isomers of conjugated linoleic acid. Prog Lipid Res 40:283–298
De la Torre A, Debiton E, Durand D, Chardigny JM, Berdeaux O, Loreau O, Barthomeuf C, Bauchart D, Gruffat D (2005) Conjugated linoleic acid isomers and their conjugated derivatives inhibit growth of human cancer cell lines. Anticancer Res 25:3943–3949
Pariza MW, Park Y, Cook ME (1999) Conjugated linoleic acid and the control of cancer and obesity. Toxicol Sci 52:107–110
Pariza MW, Park Y, Cook ME (2000) Mechanisms of action of conjugated linoleic acid: evidence and speculation. Proc Soc Exp Biol Med 223:8–13
Kelley NS, Hubbard NE, Erickson KL (2007) Conjugated linoleic acid isomers and cancer. J Nutr 137:2599–2607
Maggiora M, Bologna M, Cerù MP, Possati L, Angelucci A, Cimini A, Miglietta A, Bozzo F, Margiotta C, Muzio G, Canuto RA (2004) An overview of the effect of linoleic and conjugated-linoleic acids on the growth of several human tumor cell lines. Int J Cancer 112:909–919
Zulet MA, Marti A, Parra MD, Martínez JA (2005) Inflammation and conjugated linoleic acid: mechanisms of action and implications for human health. J Physiol Biochem 61:483–494
Gonçalves DC, Lira FS, Carnevali LC Jr, Rosa JC, Pimentel GD, Seelaender M (2010) Conjugated linoleic acid: good or bad nutrient. Diabetol Metab Syndr 30:2–62
Tian M, Kliewer KL, Asp ML, Stout MB, Belury MA (2011) c9t11-Conjugated linoleic acid-rich oil fails to attenuate wasting in colon-26 tumor-induced late-stage cancer cachexia in male CD2F1 mice. Mol Nutr Food Res 55:268–277
Graves E, Hitt A, Pariza MW, Cook ME, McCarthy DO (2005) Conjugated linoleic acid preserves gastrocnemius muscle mass in mice bearing the colon-26 adenocarcinoma. Res Nurs Health 28:48–55
McCarthy DO, Graves E (2006) Conjugated linoleic acid preserves muscle mass in mice bearing the Lewis lung carcinoma, but not the B16 melanoma. Res Nurs Health 29:98–104
Larsen AE, Crowe TC (2009) Effects of conjugated linoleic acid on myogenic and inflammatory responses in a human primary muscle and tumor coculture model. Nutr Cancer 61:687–695
Martinasso G, Maggiora M, Trombetta A, Canuto RA, Muzio G (2006) Effects of di(2-ethylhexyl) phthalate, a widely used peroxisome proliferator and plasticizer, on cell growth in the human keratinocyte cell line NCTC 2544. J Toxicol Environ Health A 69:353–365
Li G, Barnes D, Butz D, Bjorling D, Cook ME (2005) 10t,12c-conjugated linoleic acid inhibits lipopolysaccharide-induced cyclooxygenase expression in vitro and in vivo. J Lipid Res 46:2134–2142
Lee JH, Tachibana H, Morinaga Y, Fujimura Y, Yamada K (2009) Modulation of proliferation and differentiation of C2C12 skeletal muscle cells by fatty acids. Life Sci 84:415–420
Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89:381–410
Alvarez B, Quinn LS, Busquets S, López-Soriano FJ, Argilés JM (2002) TNF-alpha modulates cytokine and cytokine receptors in C2C12 myotubes. Cancer Lett 175:181–185
Bishop-Bailey D, Bystrom J (2009) Emerging roles of peroxisome proliferator-activated receptor-beta/delta in inflammation. Pharmacol Ther 124:141–150
Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Maffia P, Patel NS, Di Paola R, Ialenti A, Genovese T, Chatterjee PK, Di Rosa M, Caputi AP, Thiemermann C (2004) Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-γ, reduces acute inflammation. Eur J Pharmacol 483:79–93
Chen HH, Chen TW, Lin H (2009) Prostacyclin-induced peroxisome proliferator-activated receptor-alpha translocation attenuates NF-kappaB and TNF-alpha activation after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 297:1109–1118
Liang CJ, Tseng CP, Yang CM, Ma YH (2011) 20-Hydroxyeicosatetraenoic acid inhibits ATP-induced COX-2 expression via peroxisome proliferator activator receptor-α in vascular smooth muscle cells. Br J Pharmacol 163:815–825
Ramanan S, Kooshki M, Zhao W, Hsu FC, Robbins ME (2008) PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic Biol Med 45:1695–1704
Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B (1999) Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem 274:32048–32054
Jana M, Jana A, Liu X, Ghosh S, Pahan K (2007) Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of I kappa B alpha in anti-inflammatory effect of gemfibrozil in microglia. J Immunol 179:4142–4152
Delerive P, Gervois P, Fruchart JC, Staels B (2000) Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem 275:36703–36707
Wysong A, Couch M, Shadfar S, Li L, Rodriguez JE, Asher S, Yin X, Gore M, Baldwin A, Patterson C, Willis MS (2011) NF-κB inhibition protects against tumor-induced cardiac atrophy in vivo. Am J Pathol 178:1059–1068
Zambon A, Gervois P, Pauletto P, Fruchart JC, Staels B (2006) Modulation of hepatic inflammatory risk markers of cardiovascular diseases by PPAR-alpha activators: clinical and experimental evidence. Arterioscler Thromb Vasc Biol 26:977–986
Giordano C, Rousseau AS, Wagner N, Gaudel C, Murdaca J, Jehl-Piétri C, Sibille B, Grimaldi PA, Lopez P (2009) Peroxisome proliferator-activated receptor beta activation promotes myonuclear accretion in skeletal muscle of adult and aged mice. Pflugers Arch 458:901–913
Allen DL, Monke SR, Talmadge RJ, Roy RR, Edgerton VR (1995) Plasticity of myonuclear number in hypertrophied and atrophied mammalian skeletal muscle fibers. J Appl Physiol 78:1969–1976
Roy RR, Monke SR, Allen DL, Edgerton VR (1999) Modulation of myonuclear number in functionally overloaded and exercised rat plantaris fibers. J Appl Physiol 87:634–642
Li P, Akimoto T, Zhang M, Williams RS, Yan Z (2006) Resident stem cells are not required for exercise-induced fiber-type switching and angiogenesis but are necessary for activity-dependent muscle growth. Am J Physiol Cell Physiol 290:1461–1468
Acknowledgments
This research was supported by grants from Compagnia di San Paolo, Italy, from Regione Piemonte, Italy and from the University of Turin, Italy. We thank Frances Cooper for revising the English and Prof. Sebastiano Colombatto for technical support.
Conflict of interest
All authors have read and agreed to the editorial policies, and declare that there are no financial or other conflicts of interest that might be construed to influence the results or interpretation of their manuscript.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Oraldi, M., Maggiora, M., Paiuzzi, E. et al. CLA Reduces Inflammatory Mediators from A427 Human Lung Cancer Cells and A427 Conditioned Medium Promotes Differentiation of C2C12 Murine Muscle Cells. Lipids 48, 29–38 (2013). https://doi.org/10.1007/s11745-012-3734-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11745-012-3734-6