
Abstract
Patients with chronic obstructive pulmonary disease (COPD) are prone to clinical exacerbations that are associated with increased airway inflammation, a potent pro-thrombotic stimulus. Limited information is available on the mechanisms underlying the putative alterations of the endothelial-coagulative system during acute exacerbations. The aim was to investigate whether the activation of the endothelial-coagulative system occurs in association with the acute inflammatory response of COPD exacerbation. We monitored the blood levels of surrogate markers of inflammation: interleukin-6 (IL-6); endothelium damage: von Willebrand’s factor (vWF); clotting activation: D-dimer (D-D), and prothrombin fragment 1+2 (F1+2); fibrinolytic response: plasminogen activator inhibitor 1 (PAI-1), in COPD subjects, during hospital admission and after clinical resolution. In 30 COPD subjects, IL-6, vWF, D-D and F1+2 levels were elevated during exacerbation and decreased significantly at clinical stability (IL-6, p = 0.005; vWF, p < 0.001; D-D, p < 0.001; F1+2, p < 0.001). PAI-1 levels did not change at exacerbation compared to clinically stable situations. Positive correlations were observed between several of the markers measured. Elevation of IL-6, vWF, D-D and F1+2 levels during COPD exacerbations implies a strict association between acute inflammation, endothelial activation and clotting initiation. This was not associated with a change in PAI-1, implying an increase in the fibrinolytic response to inflammation. The pro-thrombotic nature of COPD exacerbations sustained by enhanced clotting activation appears to be mitigated by excessive fibrinolysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exacerbation of chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality [1]. COPD exacerbations are triggered by a variety of factors, including acute infections with viruses or bacteria, and are known to be associated with considerable physiological deterioration and increased airway inflammatory changes [1]. Moreover, elevated pro-inflammatory markers have been found in association with acute exacerbations of COPD (AECOPD), including the inflammatory cytokines, interleukin (IL)-6 and IL-8, the vascular activation marker, endothelin-1, and the neutrophil chemo-attractant leukotriene B4 (LTB4) [2–4]. Recent evidence indicates that the inflammation is a potent pro-thrombotic stimulus. The pro-inflammatory cytokine IL-6 is known to be critical for hemostasis not only by virtue of its ability to activate the endothelial cells, to increase tissue factor (TF) expression on the endothelium, and to enhance thrombin-induced platelet activation, but also for being a potent stimulus for the synthesis of coagulative factors such as fibrinogen and factor VIII [5]. Amplification of thrombin-induced platelet activation induced by IL-6 appears to be dependent on the endothelial release of von Willebrand factor (vWF) [6]. Hence, it is not surprising that substantial changes in the endothelial phenotype from thrombo resistant to prothrombotic is likely to occur during inflammation [7, 8]. Increased thrombin generation following clotting activation is associated with enhanced release of prothrombin fragment 1 + 2 (F1+2), and, as a result of the subsequent fibrin formation, elevated D-dimer (D-D) generation occurs [9]. Plasminogen activator inhibitor 1 (PAI-1) is the major inhibitor of the plasminogen activators, and hence it inhibits fibrinolysis [10]. Bearing in mind that expression of PAI-1 is increased by a variety of inflammatory stimuli [9, 10], changes in PAI-1 levels during AECOPD should be anticipated. A systematic review by Ambrosetti et al. [11] emphasizes that pulmonary venous thromboembolism (PTE) may be a frequent and serious threat to patients admitted for AECOPD. PTE is a blockage of the main artery of the lung or one of its branches by a substance (often a thrombus) that has travelled from elsewhere in the body through the bloodstream (embolism), whilst a venous thromboembolism (VTE) is a blood clot that forms within any vein. The clinical suspicion of PTE in patients with COPD ranges from 19 to 29% [12, 13]. However, a more recent study by Rutschmann et al. reports that PTE is probably not as prevalent in patients in the emergency department (ED) for AECOPD [14, 15]. In this study we used COPD exacerbations as a clinical model to test the hypothesis that acute inflammatory responses of COPD exacerbation are associated with the activation of the endothelial-coagulative system. This would provide a mechanism linking the acute inflammation to pro-thrombotic events in AECOPD. We have prospectively monitored the blood levels of surrogate markers of inflammation (IL-6), endothelium injury (vWF), and clotting activation (D-D and F1+2) in a cohort of patients with COPD at the time of hospital admission for disease exacerbation and after clinical resolution.
Methods
Part of the results have been previously reported as a letter-to-the-editor [16].
Study population
In this observational cohort study, we recruited 30 consecutive subjects with moderate and severe COPD with symptoms suggestive of an AECOPD admitted as emergencies to the respiratory wards. These patients were seen within 24–48 h of the onset of their symptoms, and were selected on the basis of their willingness to participate in a prospective study. The diagnosis of COPD was according to the current global initiative for chronic obstructive lung disease [17] and the patients’ smoking history was noted. The presence of COPD exacerbation was based on the criteria suggested by Anthonisen and his colleagues [18], and included an increase in all three of the symptoms of breathlessness, sputum volume and sputum purulence.
Patients with commonly acquired thrombotic risk factors including hypertension, diabetes mellitus, hypercholesterolemia, surgical procedures, malignancy and atrial fibrillation were excluded. Subjects with hypercoagulable co-morbidities (e.g. congestive heart failure, coronary heart disease) and the occurrence of critical complications of COPD exacerbations (i.e. severe acidosis and cor pulmonale) were excluded. Subjects receiving heparin, anticoagulants, statins or anti-hypertensive drugs were excluded. No changes were made to the subjects’ regular medications (including inhaled corticosteroids) in the study.
Study design
All the participants underwent an initial assessment including spirometry, arterial blood gas (ABG) and venous blood sampling. After initial assessment (admission/exacerbation; visit 1), all the patients were prescribed a standardized treatment regimen consisting of oral antibiotics, intravenous corticosteroids, nebulized bronchodilators and oxygen as recommended by current guidelines [17–19]. The possibility of significant VTE was investigated by multi-slice computed tomography (CT) scans or venous dopplers. After discharge from hospital, patients were followed up regularly to monitor the progress or deterioration by checking the changes in their symptoms and measuring regularly their peak expiratory flow (PEF) using a mini-Wright peak flow metre. Participants were asked to return for a routine follow-up appointment approximately 4 weeks after discharge (Visit 2); if their COPD was considered to be stable (i.e. when symptoms of breathlessness, sputum volume and sputum purulence recovered and morning PEF improved by at least 25% of the value obtained at visit 1), venous blood samples were taken once again. If no improvement could be demonstrated within 4 weeks after hospital discharge, the patients were considered to have unstable disease and the blood samples were not drawn. The study was approved by the Local Research Ethics Committee of the Ascoli-Tomaselli University Hospitals, and written informed consent was obtained from each patient.
Blood collection and analysis of IL-6, vWF, D-dimer, F1+2 and PAI-1
Blood samples were obtained on two different occasions: at exacerbation/admission (Visit 1; within 24–48 h of admission) and at follow-up when clinically stable (Visit 2; approximately 4 weeks after hospital discharge). Venous blood was collected into plastic tubes for serum IL-6 and then into 3.8% trisodium citrate tubes for assaying of vWF, D-D, F1+2 and PAI-1 in plasma. Samples were centrifuged, and supernatants were aliquoted and stored at −70°C until use. Serum IL-6 concentrations and plasma vWF, D-D, F1+2 and PAI-1 were measured with a enzyme-linked immunosorbent assay (ELISA) technique using commercial kits (IL-6, R&D Systems, Oxon, UK; Asserachrom vWF and PAI-1, Diagnostica Stago, Asnieres, France; Zymutest D-Dimer, Hyphen Biomed, Neuville-Sur-Oise, France¸ Enzygnost F1+2 Micro, Dade Behring; Marburg, Germany). The coefficient of variation for intra-assay variability for the measured parameters was 13, 7, 9, 11 and 8%, respectively.
Statistical analyses
Besides age, expressed as mean and standard error of the mean (SEM), all the other objective measures were not normally distributed and expressed as median values and inter-quartile range (IQR). For these variables, within group comparisons during and after clinical exacerbation of COPD were tested using Wilcoxon signed rank test. Between group analysis was conducted using univariate analysis of variance (ANOVA). Relationships between the percentage change in the parameters measured were conducted using Spearman’s rank correlation test.
Two-tailed p values <0.05 were considered to indicate statistical significance. All analyses were performed using Statistical Package for Social Sciences (SPSS Inc., Chicago, IL) for Windows version 11.5.0.
Results
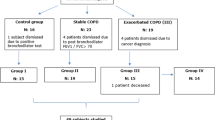
Fifty-seven (45 M,12F) consecutive patients with specifically defined COPD exacerbation criteria were initially recruited, but 23 were considered ineligible because of the exclusion criteria (Fig. 1). These patients did not differ significantly from those who remained in the study in any of the baseline parameters. Clinical deterioration requiring hospital readmission occurred in three COPD patients. Furthermore, one COPD patient was lost to follow-up. Characteristics of the remaining 30 COPD patients (27 M, 3F) are summarised in Tables 1 and 2. Neither significant VTE nor deep vein thrombosis was documented either by multi-slice computed tomography (CT) scans or by Doppler ultrasound studies of the lower limbs.

Number of patients recruited and the flow of patients within the study. Fifty-seven (45 M, 12 F) consecutive patients with specifically defined COPD exacerbation criteria were initially recruited. Twenty-three were considered ineligible because of the exclusion criteria (2 had a history of coronary heart disease; 3 having atrial fibrillation; 2 had a diagnosis of lung cancer; 5 having congestive heart failure; 11 had a combination of several conditions and/or comorbidities). Thirty-four COPD patients were able to take part in the study. However, clinical deterioration requiring hospital readmission occurred in three COPD patients. Furthermore, one COPD patient was lost to follow-up due to failure of attendance at the control visit. The remaining 30 COPD patients were available for final analyses
Functional respiratory tests and arterial blood gases
Changes in physiological parameters of COPD patients at diagnosis of an exacerbation and when clinically stable are summarised in Table 2. Compared to acute exacerbation, median forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) were measured, when COPD patients were clinically stable improved significantly by 0.1 L (from 1.04 L (0.73, 1.39) to 1.14 L (0.82, 1.58); p < 0.001) and 0.15 L (from 2.17 L (1.77, 2.60) to 2.32 L (1.81, 2.81); p < 0.001) respectively.
Compared to acute exacerbation, median PaO2 and pH (but not PaCO2) were measured, when COPD patients were clinically stable improved significantly from 59.5 (50.8, 63.0) to 67.5 (61.5, 75.0) mmHg (p < 0.001) and from 7.40 (7.33, 7.43) to 7.44 (7.42, 7.45) (p < 0.001), respectively.
Measurements of IL-6, vWF, D-D, F1+2 and PAI-1 levels
Results from the COPD study group show that IL-6, vWF, D-D and F1+2 levels are elevated during COPD exacerbations (Visit 1) and decrease significantly when patients were clinically stable (Visit 2) (Table 3), median IL-6 levels decreased from 4.95 (2.98, 9.75) to 3.2 (2.38, 4.78) pg/mL (median 35% reduction) (p < 0.001) (Fig. 2a), median vWF levels decreased from 169.6 (121.6, 231.4) to 122 (91.1, 160.2) pg/mL (median 28% reduction) (p < 0.001) (Fig. 2b), median D-D levels decreased from 157.5 (141.75, 233) to 127 (111.25, 151.5) ng/mL (median 19% reduction) (p < 0.001) (Fig. 2c), median F1+2 levels decreased from 1.19 (0.95, 1.66) to 0.73 (0.53, 0.91) nmol/L (median 39% reduction) (p < 0.001) (Fig. 2d).

Changes in individual serum a IL-6 concentrations (IL-6: 0.44–8.50 pg/ml), b vWF concentrations (vWF: 50–140 pg/ml), c D-Dimer concentrations (D-D: below 200 ng/ml), d F1+2 concentrations (F1+2: 0.2–0.6 nmol/L), e PAI-1 concentrations (PAI-1: 4.0–30.0 ng/ml) obtained from 30 COPD patients during an acute exacerbation and when clinically stable. Median values are shown as horizontal bars. Dotted lines delineate reference values for healthy donors
No statistically significant changes of PAI-1 levels were observed during COPD exacerbations in comparison with clinically stable situations, their median levels being 11.35 (9.38, 14.28) and 10.7 ng/mL (8.48, 13.35) (p = 0.765), respectively (Fig. 2e).
Correlations among pro-thrombotic, fibrinolytic and inflammatory markers
Statistical analysis revealed a positive correlation between the percentage change in concentrations of IL-6 and D-D (ρ = 0.56, p < 0.001); percentage change in concentrations of D-D levels and vWF (ρ = 0.38, p < 0.05); and the percentage change in concentrations of PAI-1 and vWF (ρ = −0.415, p = 0.022).No other significant associations were observed for all the other parameters measured in the blood.
Moreover, statistical analysis also failed to reveal any significant association during acute exacerbation and at clinical stability between the percentage change for all physiological parameters measured and in the blood levels for IL-6, vWF, D-D and F1+2 during COPD exacerbations.
Discussion
This study provides evidence that a pro-thrombotic condition is associated with AECOPD. We have shown for the first time that the levels of vWF, D-D and F1+2 are elevated in COPD patients during acute exacerbation of their disease, and decline substantially when clinically stable. In addition, IL-6, a critical inflammatory cytokine with pro-thrombotic effects, is elevated during acute exacerbation. Positive correlations between IL-6 and D-D and between vWF and D-D changes during exacerbation are also observed.
Taken together these observations imply a strict association between acute inflammation, and endothelial and clotting activation. The data of the present study are in agreement with those of others. Earlier work by Wedzicha et al. [20] postulates an enhanced platelet activity in COPD by showing lower platelet aggregate ratio in hypoxemic COPD patients compared to controls with a trend to lesser aggregate ratios in the more hypoxemic patients. Further support to the view that a pro-thrombotic condition is present in COPD was provided by Alessandri et al. [21], who demonstrated that about two-thirds of patients with stable COPD have elevated the plasma levels of the thrombin generation marker F1+2. The results of the study show an increase of IL-6 levels during COPD exacerbations as compared with the levels during stable conditions, these data are in agreement with previous work showing that COPD patients have increased the blood level of fibrinogen and IL-6 during exacerbations of their disease [22].
Smoking per se may be responsible for the observed endothelial/platelet dysfunction and clotting activation. There is sound evidence that cigarette smoking is associated with high levels of vWF [23]. Moreover, we have reported that smoking cessation reduces vWF and D-D plasma levels in regular smokers [24]. However, when the effects of AECOPD on the blood levels of IL-6, vWF, and D-D levels are compared between COPD smokers and never smokers, we observe a similar decline in IL-6, vWF, and D-D levels during clinical stability. That a pro-thrombotic condition is present in COPD patients regardless of their smoking status has also been reported [21].
The hypoxia associated with an AECOPD is another factor that may induce the high levels of vWF, D-D, and F1+2. Basic science and animal studies indicate that hypoxia in isolation may elicit a pro-thrombotic state [25], and enhances pro-coagulant activity of endothelial cells in vitro [26, 27]. An uncontrolled study in healthy subjects shows that acute induction to hypoxia increases factor VIII in the blood [28]. However, when the effects of AECOPD on the blood levels of vWF and D-D levels were analysed for statistical association with the percentage change in PaO2, we failed to show any correlation of statistical relevance. This is in agreement with the notion that it has been hard to prove that hypoxia is a pro-thrombotic stimulus in man either in altitude chambers or in field studies [29].
A large variety of clotting and inflammation biomarkers including fibrinogen, anti-thrombin III (AT III), protein C (PC), protein S (PS), activated protein C ratio (APCR), P-selectin, E-selectin and L-selectin [30, 31] could have been also assessed. During exacerbations, patients with COPD, compared to controls, have elevated the serum levels of fibrinogen and P-selectin. In particular, circulating P-selectin, which is crucial for platelet aggregation and platelet-leucocyte interactions [32] is negatively correlated to low arterial oxygen tensions. Furthermore, there are reductions in the natural anti-coagulants AT III, PS, APCR and PC during disease exacerbation, however only the latter to significantly low levels. Systemic steroids have been reported to significantly attenuate serum CRP, IL-6 and fibrinogen levels [30, 33], and increase AT III, PC and PS levels [30]. Although systemic steroids have been implicated in improving the hypercoagulable state, blood gases, lung function and restoring hemodynamic states, other therapeutic options such as anti-aggregate, selectin inhibitors and anti-thrombins need to be further investigated [30, 34].
The clinical implication of elevated vWF, D-Dimer and F1+2 concentrations in the blood during AECOPD is that this condition might increase the likelihood of thromboembolic events in these patients just as it has been reported in ischemic heart disease [35, 36]. Death from PTE occurs in about 10% of patients admitted for an AECOPD [37]. Numerous studies report incidences of acute VTEs as high as 31% during admission for AECOPD [13, 38, 39]. However, the prevalence of PTE was questioned when in a recent study only 6.2% of suspected and 1.3% of unsuspected patients who attended the ED for an AECOPD were diagnosed as having PTE [14]. Concerns were raised of the prior studies having a selection bias of patients. Suggestions have been raised that prolonged and complicated hospital stays may have higher incidences of VTEs and that patients having re-admissions secondary to further exacerbation recurrences may be attributed to PTE [15].
Although an increase in PAI-1 levels during COPD exacerbations was expected, this was not observed in the present study. The absence of changes in PAI-1 levels suggests that an enhanced fibrinolytic response to inflammatory stimuli may occur. This observed lack of elevation in PAI-1 levels may be an endogenous protective mechanism that attenuates the pro-thrombotic state in the course of COPD exacerbations. This may explain the lack of clinically relevant thrombotic events observed in the patients studied, as has been reported previously [14].
There are a number of limitations in our study. First, some of the patients in the study group were not very severe in terms of their COPD exacerbations, hence appropriate characterisation of the patients in terms of their severity may have provided a better idea about the risk of PTE. However, we did assess their PEF and symptoms at recovery and a 30% increment in PEF from the time of exacerbations is an indication of reasonably severe AECOPD, we concede that systemic steroids may influence early clotting activation results. However, blood was collected up to 4 weeks after hospital discharge, and during this period no systemic steroid was given. Therefore, given the known pharmacodynamic of IV methylprednisolone, it is unlikely that the effect observed in this study may be due to a direct inhibition caused by the steroid treatment [40].
The transient elevation in IL-6, vWF, D-D and F1+2 levels in association with COPD exacerbation suggest a composite relationship between acute inflammation, endothelial activation and clotting initiation. The heightened clotting state during COPD exacerbation may predispose to venous thromboembolism (as in acute ischemic heart disease) and could, in principle, justify a general recommendation for anticoagulation and pharmacological thrombo prophylaxis in these patients during exacerbation of their disease. Yet, only direct visualization of ongoing thrombosis by multi-slice CT scans and Doppler ultrasonography in prospective studies will establish realistic risk levels for pulmonary emboli and/or DVT and confer clinical relevance to our observations.
References
Wedzicha JA, Donaldson GC (2003) Exacerbations of chronic obstructive pulmonary disease. Respir Care 48(12):1204–1213 discussion 1213–1205
Roland M, Bhowmik A, Sapsford RJ, Seemungal TA, Jeffries DJ, Warner TD, Wedzicha JA (2001) Sputum and plasma endothelin-1 levels in exacerbations of chronic obstructive pulmonary disease. Thorax 56(1):30–35
Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA (2000) Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 55(2):114–120
Montuschi P, Sala A, Dahlen SE, Folco G (2007) Pharmacological modulation of the leukotriene pathway in allergic airway disease. Drug Discov Today 12:404–412
Kerr R, Stirling D, Ludlam CA (2001) Interleukin 6 and haemostasis. Br J Haematol 115(1):3–12
Newby DE, Stewart A, Witherow FN, Grieve S, Dawson P, Fox KA, Webb DJ, Ludlam CA (2000) Local and systemic effects of intra-arterial desmopressin in healthy volunteers and patients with type 3 von Willebrand disease. Role of interleukin-6. Thromb Haemost 84(2):195–203
Cirino G, Napoli C, Bucci M, Cicala C (2000) Inflammation-coagulation network: are serine protease receptors the knot? Trends Pharmacol Sci 21(5):170–172
Esmon CT (2003) Inflammation and thrombosis. J Thromb Haemost 1(7):1343–1348
Dahlback B (2000) Blood coagulation. Lancet 355(9215):1627–1632
Salgado A, Boveda JL, Monasterio J, Segura RM, Mourelle M, Gomez-Jimenez J, Peracaula R (1994) Inflammatory mediators and their influence on haemostasis. Haemostasis 24(2):132–138
Ambrosetti M, Ageno W, Spanevello A, Salerno M, Pedretti RF (2003) Prevalence and prevention of venous thromboembolism in patients with acute exacerbations of COPD. Thromb Res 112(4):203–207
Hartmann IJ, Hagen PJ, Melissant CF, Postmus PE, Prins MH (2000) Diagnosing acute pulmonary embolism: effect of chronic obstructive pulmonary disease on the performance of D-dimer testing, ventilation/perfusion scintigraphy, spiral computed tomographic angiography, and conventional angiography. ANTELOPE Study Group. Advances in New Technologies Evaluating the Localization of Pulmonary Embolism. Am J Respir Crit Care Med 162(6):2232–2237
Tillie-Leblond I, Marquette CH, Perez T, Scherpereel A, Zanetti C, Tonnel AB, Remy-Jardin M (2006) Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: prevalence and risk factors. Ann Intern Med 144(6):390–396
Rutschmann OT, Cornuz J, Poletti PA, Bridevaux PO, Hugli OW, Qanadli SD, Perrier A (2007) Should pulmonary embolism be suspected in exacerbation of chronic obstructive pulmonary disease? Thorax 62(2):121–125
Wedzicha JA, Hurst JR (2007) Chronic obstructive pulmonary disease exacerbation and risk of pulmonary embolism. Thorax 62(2):103–104
Polosa R, Cacciola RR, Prosperini G, Spicuzza L, Morjaria JB, Di Maria GU (2008) Endothelial-coagulative activation during chronic obstructive pulmonary disease exacerbations. Haematologica 93(8):1275–1276
Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS (2001) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 163(5):1256–1276
Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA (1987) Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 106(2):196–204
Montuschi P (2006) Pharmacological treatment of chronic obstructive pulmonary disease. International Journal of Chronic Obstructive Pulmonary Disease 1:409–423
Wedzicha JA, Syndercombe-Court D, Tan KC (1991) Increased platelet aggregate formation in patients with chronic airflow obstruction and hypoxaemia. Thorax 46(7):504–507
Alessandri C, Basili S, Violi F, Ferroni P, Gazzaniga PP, Cordova C (1994) Hypercoagulability state in patients with chronic obstructive pulmonary disease. Chronic Obstructive Bronchitis and Haemostasis Group. Thromb Haemost 72(3):343–346
Wedzicha JA, Seemungal TA, MacCallum PK, Paul EA, Donaldson GC, Bhowmik A, Jeffries DJ, Meade TW (2000) Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost 84(2):210–215
Blann AD, McCollum CN, Lip GY (2002) Relationship between plasma markers of endothelial cell integrity and the Framingham cardiovascular disease risk-factor scores in apparently healthy individuals. Blood Coagul Fibrinolysis 13(6):513–518
Caponnetto P, Russo C, Di Maria A, et al. (2010) Circulating endothelial-coagulative activation markers after smoking cessation: a 12-month observational study. Eur J Clin Invest Dec 29. [Epub ahead of print]
Yan SF, Lu J, Zou YS, Soh-Won J, Cohen DM, Buttrick PM, Cooper DR, Steinberg SF, Mackman N, Pinsky DJ et al (1999) Hypoxia-associated induction of early growth response-1 gene expression. J Biol Chem 274(21):15030–15040
Gertler JP, Weibe DA, Ocasio VH, Abbott WM (1991) Hypoxia induces procoagulant activity in cultured human venous endothelium. J Vasc Surg 13(3):428–433
Ogawa S, Shreeniwas R, Brett J, Clauss M, Furie M, Stern DM (1990) The effect of hypoxia on capillary endothelial cell function: modulation of barrier and coagulant function. Br J Haematol 75(4):517–524
Le Roux G, Larmignat P, Marchal M, Richalet JP (1992) Haemostasis at high altitude. Int J Sports Med 13(Suppl 1):S49–S51
Grover RF, Bartsch P (2002) High altitude: an exploration of human adaptation. Marcel Dekker, New York
Kunter E, Ilvan A, Ozmen N, Demirer E, Ozturk A, Avsar K, Sayan O, Kartaloglu Z (2008) Effect of corticosteroids on hemostasis and pulmonary arterial pressure during chronic obstructive pulmonary disease exacerbation. Respiration 75(2):145–154
Cella G, Sbarai A, Mazzaro G, Vanzo B, Romano S, Hoppensteadt T, Fareed J (2001) Plasma markers of endothelial dysfunction in chronic obstructive pulmonary disease. Clin Appl Thromb Hemost 7(3):205–208
Yokoyama S, Ikeda H, Haramaki N, Yasukawa H, Murohara T, Imaizumi T (2005) Platelet P-selectin plays an important role in arterial thrombogenesis by forming large stable platelet-leukocyte aggregates. J Am Coll Cardiol 45(8):1280–1286
Sin DD, Lacy P, York E, Man SF (2004) Effects of fluticasone on systemic markers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 170(7):760–765
Davies L, Angus RM, Calverley PM (1999) Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: a prospective randomised controlled trial. Lancet 354(9177):456–460
Lowe GD, Yarnell JW, Sweetnam PM, Rumley A, Thomas HF, Elwood PC (1998) Fibrin D-dimer, tissue plasminogen activator, plasminogen activator inhibitor, and the risk of major ischaemic heart disease in the Caerphilly Study. Thromb Haemost 79(1):129–133
Rumley A, Lowe GD, Sweetnam PM, Yarnell JW, Ford RP (1999) Factor VIII, von Willebrand factor and the risk of major ischaemic heart disease in the Caerphilly Heart Study. Br J Haematol 105(1):110–116
Groenewegen KH, Schols AM, Wouters EF (2003) Mortality and mortality-related factors after hospitalization for acute exacerbation of COPD. Chest 124(2):459–467
Erelel M, Cuhadaroglu C, Ece T, Arseven O (2002) The frequency of deep venous thrombosis and pulmonary embolus in acute exacerbation of chronic obstructive pulmonary disease. Respir Med 96(7):515–518
Mispelaere D, Glerant JC, Audebert M, Remond A, Sevestre-Pietri MA, Jounieaux V (2002) [Pulmonary embolism and sibilant types of chronic obstructive pulmonary disease decompensations]. Rev Mal Respir 19(4):415–423
Willaert W, Daenen M, Bomans P, Verleden G, Decramer M (2002) What is the optimal treatment strategy for chronic obstructive pulmonary disease exacerbations? Eur Respir J 19(5):928–935
Acknowledgments
This study was supported in part by a personal research grant to R.P. from the University of Catania (Grant 60%). RP, MM, RRC and JBM have contributed equally to the work.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Polosa, R., Malerba, M., Cacciola, R.R. et al. Effect of acute exacerbations on circulating endothelial, clotting and fibrinolytic markers in COPD patients. Intern Emerg Med 8, 567–574 (2013). https://doi.org/10.1007/s11739-011-0636-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-011-0636-1