
Abstract
Acute hyperglycemia frequently present in stress conditions, has long been generally accepted as normal, and not thought to be a cause for concern since a moderate hyperglycemia in critically ill adult patients has been thought to be beneficial during the “fight or flight” response to ensure a supply of glucose as a source of energy to organs that do not require insulin for glucose uptake (i.e., the brain and the immune system). However, an increasing body of evidence associates the upon-admission degree and duration of hyperglycemia during critical illness with an adverse outcome. Hyperglycemia should be regarded as a part of the systemic and complex metabolic derangements observed in critical illness in response to stress and inflammation, which can lead, independent of initial disease, to multiorgan dysfunction and death. A tight glycemic control should be constantly pursued and achieved by insulin infusion bearing in mind that the therapeutic target is fighting the systemic inflammatory response and not merely the glucose plasma levels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the past decades, the evolution of critical care medicine has fostered a tremendous increase in the immediate survival of acutely ill patients who frequently enter a “chronic phase” of critical illness during which they are still dependent on vital organ support for quite a long period. In these patients, mortality remains high, with a 20% risk of death, mostly due to non-resolving multiple organ failure, regardless of the initial disease. In this phase, continuing hypercatabolism results in profound erosion of lean body mass despite artificial feeding and relative preservation of adipose tissue. Among other metabolic and endocrine abnormalities, these patients usually develop hyperglycemia, reflecting insulin resistance and accelerated glucose production, known as stress diabetes or the diabetes of injury [1, 2].
Acute hyperglycemia, frequently present in stress conditions [3, 4], has long been generally accepted as non-problematic, since a moderate hyperglycemia in critically ill adult patients has been thought to be beneficial during the “fight or flight” response to ensure the supply of glucose as a source of energy to organs that do not require insulin for glucose uptake (i.e. the brain and the immune system). However, an increasing body of evidence associates the upon-admission degree of hyperglycemia and the duration of hyperglycemia during critical illness with an adverse outcome. The important issue is whether hyperglycemia is just related to disease severity or is an independent risk factor that contributes to morbidity and mortality in critically ill adult patients [5].
Glucose metabolism and critically illness
In physiological conditions, despite large timely fluctuations in supply and demand, plasma glucose levels are normally controlled within a narrow range between 80 and 125 mg/dl in a fasting state. Interestingly, 80% of systemic glucose utilization occurs by non-insulin-mediated glucose uptake under basal conditions, mainly by the central nervous system [6]. Glucose transport into cells occurs as facilitated diffusion using one of the five glucose transported (GLUT) channel proteins (GLUT1-mediated insulin independent transport mostly in basal conditions; GLUT4 specifically and reversibly up regulated by insulin). During moderate hyperglycemia, cells usually respond with a removal of GLUT transport molecules to protect themselves from glucose overload [7].
In the critically ill patient, the classic endocrine reaction to a stressful challenge consists of the activation of the sympathoadrenal and the hypothalamopituitary-adrenal axis, leading to increased plasma levels of catecholamines and glucocorticoids [1], and the degree of systemic response to stress correlates with the intensity of the challenge (Table 1).
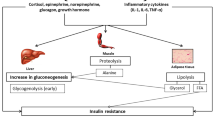
Stress, imposed by any type of acute illness, leads to the development of insulin resistance, glucose intolerance and hyperglycemia [8] (Fig. 1). In the acute phase of critical illness, despite high plasma glucose levels and abundantly released insulin, hepatic glucose production is upregulated probably due to elevated levels of cytokines, growth hormone, glucagons and cortisol [2, 9]. Hepatic glycogenolysis is further enhanced by catecholamines that also inhibit glycogen-neogenesis [10]. Moreover, glucose uptake mechanisms are also affected in the critically ill and contribute to hyperglycemia. Exercise-stimulated glucose uptake in skeletal muscle totally disappears because of immobilization [11]. Glucose uptake in heart, skeletal muscle, and adipose tissue is compromised because of impaired insulin-stimulated glucose uptake by the glucose transporter 4 (GLUT-4) and impaired glycogen synthetase [8]. Nevertheless, it has been observed that the total body glucose uptake is massively increased, but is accounted for by tissues that do not depend on insulin for glucose uptake such as brain and blood cells [1]. The higher levels of insulin, the impaired peripheral glucose uptake and the elevated hepatic glucose production reflect the development of an insulin resistance during critical illness.

The relationship between stress and hyperglycemia in the critically ill
Interestingly, in critically ill patients, hyperglycemia seems more acutely toxic than in healthy individuals (in whom cells can protect themselves by down-regulating glucose transporters [7]. There are two possible explanations, not mutually exclusive, for this phenomenon [12]. The first is the accentuated cellular glucose overload, and the second is the more pronounced toxic side effects of glycolysis and oxidative phosphorylation.
Cellular glucose overload
The central and peripheral nervous system, hepatocytes, endothelial, epithelial, and immune cells take up glucose independently of insulin. Three glucose transporters, GLUT-1, GLUT-2, and GLUT-3, facilitate insulin-independent glucose transport in these tissues. In physiological conditions, hyperglycemia induces a down-regulation in GLUT-1 transporters, thus protecting cells against glucose overload. Cytokines, growth factors and hypoxia all increased in critical illness have been shown to up-regulate expression of the glucose transporters (GLUT-1, GLUT2 and GLUT3), thus overriding the physiological down-regulatory protective response against hyperglycemia in several cellular types (hepatocytes, gastro-intestinal mucosal cells, pancreatic beta-l cells, renal tubular cells, endothelial cells, immune cells and neurons, all insulin independent for glucose uptake) [13]. Hence, particularly in critical illness, characterized by high circulating levels of all these regulators, all organ systems that take up glucose passively may theoretically be at high risk for direct glucose toxicity. In contrast, skeletal muscle and the myocardium, which normally take up glucose predominantly via the insulin-dependent GLUT-4 transporter, may be relatively protected against toxic effects of circulating glucose.
Increased oxidative phosphorylation in the critically ill
Vulnerability to glucose toxicity may be also due to increased generation of or deficient scavenging systems for radical oxygen species (ROS) produced by activated glycolysis and oxidative phosphorylation [14]. In physiological conditions, glucose in the cytosol undergoes glycolysis, and its metabolite pyruvate is further transformed into acetyl-CoA, after which, in the presence of O2, oxidative phosphorylation generates ATP. Along with the generation of ATP by the mitochondrial respiratory chain complexes I–V, a small amount of superoxide is concomitantly produced. In physiological conditions, 2–5% of O2 used in the mitochondria is metabolized into superoxide, which is subsequently detoxified by manganese superoxide dismutase (MnSOD). When more glucose enters the cell and more pyruvate is being used for oxidative phosphorylation, more superoxide will be generated [13]. Superoxide interacts with NO to form peroxynitrite, which nitrates several protein substrates such as mitochondrial complexes I and IV and MnSOD. During critical illness, hypoxia-reperfusion aggravates superoxide production and when, in this setting, cells are also overloaded with glucose, high levels of superoxide (together with nitric oxide metabolites) contribute to mitochondrial dysfunction (by inhibiting the glycolytic enzyme GAPDH and mitochondrial complexes I and IV). Liver biopsies reveal profound ultrastructural abnormalities in hepatocytic mitochondria of patients with moderate hyperglycemia in whom conventional insulin therapy is randomly administered, while no abnormalities are found in patients submitted to intensive insulin therapy [15]. The higher activity of respiratory chain complex I and complex IV can be considered the functional correlate of the prevention or reversal of these morphological abnormalities in hepatocytic mitochondria. Conversely, no major ultrastuctural abnormalities are found in the mitochondria of skeletal muscle, thus suggesting that in this tissue, insulin therapy is not able to affect the respiratory chain activity. This phenomenon strongly suggests a direct effect of glucose control, rather than of insulin. In those tissues (such as immune and endothelial cells, the central and peripheral nervous system) where glucose enters passively, reduction in plasma glucose levels would probably lead to prevention of hyperglycemia-induced mitochondrial dysfunction, thus explaining some of the protective effects of insulin therapy in critically ill patients. In fact, in the Leuven study [16], it is observed that maintaining normoglycemia with insulin is associated with a reduced incidence of severe nosocomial infections and lethal sepsis. Protection of the peripheral nervous system is evidenced by prevention of the development of critical illness polyneuropathy, which results in a shorter duration of mechanical ventilation and intensive care stay, while the observed reduced incidence of seizures [17] can be related to a more efficacious protection of the central nervous system.
At the cellular level, severe hyperglycemia has been found to induce repeated and acute changes in cellular metabolism and in the structure of macromolecules [14, 18]. In the presence of high glucose concentrations, several steps of the glycolytic pathways can induce the release of toxic derivatives, including polyols derived from glucose, hexosamines from fructose-6-phosphate, advanced glycation products, and activators of the protein kinase C pathway from glyceraldehyde-3-phosphate.
Prevalence and prognostic value of hyperglycemia
A correlation between hyperglycemia and mortality has been described in varying situations [19], even though different authors use different thresholds to define hyperglycemia.
Among patients admitted to a general hospital [20], hyperglycemia (defined as fasting blood glucose above 7 mmol/l to >140 mg/dl)is observed in 38%. Among the patients with newly discovered hyperglycemia, 16% die during hospital stay, as compared with only 1.7% of patients with normoglycemia (P < 0.001). Patients with new onset hyperglycemia are more often admitted to the ICU (29 vs. 19%) and have a longer hospital stay. In this investigation, known diabetic patients show a better prognosis than newly hyperglycemic patients.
In medical ICU patients [21] hyperglycemia is present in 23% of patients, while it is detectable in 86% of thoracosurgical ICU patients [16]; most of them (96%) become hyperglycemic during their ICU stay.
Hyperglycemia is a risk factor for increased morbidity and mortality in critically ill surgical patients [22] but not in medical ones, though the number of medical patients enrolled in this study was small, and thus no firm conclusions can be drawn. In different ICU populations, data on the association between hyperglycemia and in-hospital mortality are controversial.
In a retrospective study [23] performed in a mixed ICU population, even a modest degree of hyperglycemia is associated with an increased in-hospital mortality, whereas in another retrospective study [5] hyperglycemia is not an independent risk factor for mortality in a multivariate model.
Whitcob et al. [24] assess, in a retrospective cohort study, the generalizability of the association between hyperglycemia and in-hospital mortality in different intensive care unit types adjusting for illness severity and diabetic history. Admission blood glucose is used to classify patients as hyperglycemic (>200 mg/dL) or normoglycemic (60–200 mg/dL). The adjusted odds ratios for death comparing all patients with hyperglycemia to all those without are 0.81 (95% confidence interval, 0.37, 1.77) and 1.76 (95% confidence interval, 1.23, 2.53) for those with and without a diabetic history, respectively. A higher mortality is seen in hyperglycemic patients without a diabetic history in the cardiothoracic, (adjusted odds ratio, 2.84 [1.21, 6.63]), cardiac (adjusted odds ratio, 2.64 [1.14, 6.10]), and neurosurgical units (adjusted odds ratio, 2.96 [1.51, 5.77]), but not in the medical or surgical intensive care units, or in patients with a diabetic history. The authors conclude that the association between hyperglycemia on intensive care unit admission and in-hospital mortality is not uniform in the study population, and that hyperglycemia is an independent risk factor only in patients without a diabetic history in the cardiac, cardiothoracic, and neurosurgical intensive care units. Similarly Wahab et al. [25] document a two-fold increase in odds ratio for mortality in hyperglycemic non-diabetic patients, which is higher compared to the risk observed in diabetic patients. The reason that critically ill patients with a history of diabetes may not have the same degree of hyperglycemia-associated mortality as non-diabetic patients is not clear. It can be hypothesized that patients with a history of diabetes are more likely to receive intensive insulin treatment during their stay. In a recent paper by Giannazzo et al. [26] (and in the Editorial by Ban et al. [27]), in a retrospective study performed in patients admitted to the ED with a diagnosis of severe sepsis or septic shock, some therapy with insulin is the only variable related to a lower risk of death and, at univariate analysis, a history of diabetes predicts a favorable outcome. This phenomenon may be related to the fact that diabetics are treated earlier and much more aggressively since they are easily recognized as being at a higher risk of sepsis. Diabetic patients represent a unique patient population at higher risk for complications. Myocardial infarction is an uncommon but well recognized precipitant of diabetic ketoacidosis (DKA) accounting for 1% of cases. Myocardial infarction, and congestive cardiac failure account for 28% of deaths in DKA [28]. In a recent investigation [29], it is observed that elevated troponin I in diabetic patients admitted with DKA identifies, though in the absence of clear acute coronary syndrome, a group at very high risk for future cardiac events and mortality. The triad of uncontrolled hyperglycemia, metabolic acidosis and increased total body ketone concentration characterizing DKA result from the combination of absolute or relative insulin deficiency and increased levels of counter-regulatory hormones (glucagon, catecholamines, cortisol, and growth hormone). All these factors taken together can contribute to the development of myocardial ischemia together with fluid shift, tachycardia, and increased sympathetic tone.
In patients with acute myocardial infarction (AMI) [30], mild or moderate impaired glucose levels (5–8 mmol/l to 100–160 mg/dl) are observed in 65% of patients while previously known diabetes is present in 15%. This investigation, performed in an unselected population of AMI patients, documents that admission glucose level is independently correlated with 1-year mortality, thus underlying the prognostic importance of impaired glucose in these patients. In a meta-analysis of 1,856 AMI patients [31], hyperglycemia is associated with an increased risk of in-hospital mortality in patients with and without diabetes; the risk of congestive heart failure or cardiogenic shock is increased in patients without diabetes. In a retrospective study with prospective follow-up performed in 846 AMI patients [32], the admission blood glucose level after AMI is confirmed as an independent predictor of long-term mortality in patients with and without known diabetes. The AMI patients with unknown diabetes and admission glucose levels of 200 mg/dL or more show mortality rates comparable to those with established diabetes. In 779 consecutive AMI patients from the Myocardial Infarction Registry in Estonia [33] hyperglycemia on admission, independent of a history of diabetes, represents an independent risk factor for 180-day mortality; non-diabetic hyperglycemic patients experience the worst outcome. Aaronson et al. [34] prospectively studied the relationship between fasting glucose level and long-term mortality in AMI patients, and find a graded independent association between glucose levels at admission and long-term mortality in non-diabetic patients. Both admission and fasting glucose levels provide incremental prognostic information with regard to long-term mortality when added to the GRACE risk score. However, fasting glucose provides greater incremental prognostic information than admission glucose. Furthermore, fasting glucose remains an independent predictor of long-term mortality after adjustment for left ventricular ejection fraction. Recently also in patients with acute coronary syndrome [35], an elevated glucose level on admission is a significant independent predictor of in-hospital mortality, and is even more important in patients who do not have known diabetes.
In a retrospective study in patients with an established cerebrovascular accident (CVA) [36] admitting hyperglycemia is common (about 40%), and is associated with increased short- and long-term mortality and with increased inpatient charges. In CVA patients without known diabetes but with an elevated blood glucose (>6.1 mmol/l to >122 mg/dl) [37], the risk of dying within 30 days is threefold higher than in the normoglycemic patients.
In trauma patients [38] even a mild hyperglycemia (>7.5 mmol/l to >150 mg/dl) is an independent predictor for mortality, postoperative infection and hospital and ICU length of stay. The relationship between hyperglycemia and mortality in these patients is more pronounced than in elective surgical patients [38]. It has been described that in trauma patients the elevated blood sugar, in the early phase, results from glycogen mobilization, while hyperglycemia in the late phase (4–5 days after injury) develops if organ failure or infectious complications ensue. Patients with hyperglycemia on admission have higher mortality rates, whereas blood glucose levels at 24 h do not correlated with outcome, particularly if the patient is adequately resuscitated with a normal lactate level [39]. As far as therapy is concerned, despite the increased use of insulin drips and the higher number of glucose checks after adopting a stricter insulin treatment protocol, ICU outcomes remained unchanged [40]. In an experimental model of activated macrophages [41], insulin does not prove to have direct anti-inflammatory properties.
On the basis of available literature data, the incidence of admission hyperglycemia in critically ill patients is quite high, and in the vast majority of the studies, inpatient hyperglycemia is associated with a lower survival rate [19, 42]. Many of the currently available studies are limited because they do not adequately control for severity of illness, they focus only on specific subsets of patients, or they define hyperglycemia by admission glucose only. However, these observational studies have been informative in areas where randomized trials are difficult or impractical.
Insulin and the critically ill
Benefits of insulin therapy in critically illness
The Leuven Study [16], a prospective, randomized, controlled investigation of a large group of patients (the majority of whom did not previously have diabetes) shows that titrating insulin infusion during intensive care to strict normoglycemia (below 110 mg/dl) strikingly reduces mortality (43% reduction) when compared with conventional insulin treatment. Besides, intensive insulin therapy largely prevents critical associated complications; the incidence of critical illness polyneuropathy is reduced by 44%, the development of bloodstream infections by 46% and acute renal failure requiring dialysis or hemofiltration by 41%. Patients are also less dependent on mechanical ventilation. The clinical benefits of this regimen are also observed in most diagnostic subgroups such as patients with isolated brain injury [43].
The clinical benefits of insulin therapy are also observed in a heterogeneous medical and surgical population in a non-randomized setting [16, 44]. In a prospective, randomized, controlled study of adult medical patients admitted to ICU [3], it is clearly demonstrated that insulin therapy reduces morbidity and mortality of patients treated at least a few days in the ICU. A higher number of deaths are documented in the group of short-stay patients on intensive insulin therapy, and although this difference is not statistically significant and likely explained by selection bias, it is of concern to the practicing clinician.
Pooling the two data sets of randomized controlled trials [16, 45] generates equal-sized samples of long-stay and short-stay medical/surgical ICU patients. Intensive insulin therapy significantly reduces morbidity and mortality in mixed medical/surgical ICU patients in an intention-to-treat analysis. This effect is increased when the therapy is continued for at least 3 days, independent of parenteral glucose load, and without causing harm to patients treated for <3 days. Patients with a prior history of diabetes do not appear to show benefit, probably because in these patients a rapid normalization of blood glucose levels can be deleterious especially if their blood glucose levels before ICU admission are elevated. [45].
Other studies on insulin intensive therapy in different patients’ subsets
In patients with AMI, the infusion of glucose together with insulin and potassium (GIK) is associated with a reduction in early mortality and morbidity, thus yielding promising results [46]. However, recently two large, randomized trials on GIK therapy in AMI patients (CREATEECLA [Clinical Trial of Reviparin and Metabolic Modulation in Acute Myocardial Infarction Treatment and Evaluation: Estudios Cardiologicas Latin America]) and in AMI patients with diabetes and myocardial infarction (DIGAMI-2, [second Diabetes and Insulin–Glucose Infusion in Acute Myocardial Infarction study]) do not show clinical benefits of this treatment [46, 47]. In the CREATE-ECLA study, the patients who received GIK and those who did not show a comparable mortality and incidence of cardiac arrest, cardiogenic shock, and reinfarction [47]. However, no simultaneous glucose control was performed, and GIK therapy was associated with an increase in glucose levels even compared with the usual care. The DIGAMI-2 trial [48] was set up to investigate whether the clinical benefit of the first DIGAMI study [49–51] was mediated by an acute effect of GIK and that of blood glucose control during the months after the infarction. This study observes no significant effect on morbidity and mortality, assessed by the occurrence of non-fatal reinfarctions and strokes. However, the target of normal glucose levels assigned to one of the groups was not reached, thus excluding an acute effect of GIK in the absence of blood glucose control. Therefore, no conclusion can be drawn with respect to the effect of blood glucose control because of unintended protocol violation.
Similarly, GIK infusion for 24 hrs following acute stroke does not realize a significant reduction in glycemia or mortality, as reported in the Glucose–Insulin in Stroke Trial (GIST) [52]. Recently [53] in a multicenter, two-by-two factorial trial, patients with severe sepsis were randomly assigned to receive either intensive insulin therapy to maintain euglycemia or conventional insulin therapy and either 10% pentastarch, a low-molecular-weight hydroxyethyl starch (HES 200/0.5), or modified Ringer’s lactate for fluid resuscitation. The rate of death at 28 days and the mean score for organ failure were coprimary end points. The trial was stopped early for safety reasons. Among 537 patients who could be evaluated, the mean morning blood glucose level is lower in the intensive-therapy group (112 mg per deciliter [6.2 mmol per liter]) than in the conventional-therapy group (151 mg per deciliter [8.4 mmol per liter], P < 0.001). However, at 28 days, there is no significant difference between the two groups in the rate of death or the mean score for organ failure. The rate of severe hypoglycemia (glucose level, ≤40 mg per deciliter [2.2 mmol per liter]) is higher in the intensive-therapy group than in the conventional-therapy group (17.0 vs. 4.1%, P < 0.001), as is the rate of serious adverse events (10.9 vs. 5.2%, P = 0.01). The HES therapy is associated with higher rates of acute renal failure and renal-replacement therapy than is Ringer’s lactate.
In “real-life” intensive care of a heterogeneous critically ill medical/surgical patient population, the clinical benefits of implementing a tight glucose management protocol, by comparison with historical controls as a reference are confirmed in an observational study [44] Intravenous insulin was administered only if glucose levels exceeded 200 mg/dL on two successive measurements and aimed to lower glycemia to 140 mg/dL, (which is less strict than in the Leuven studies). When compared to a historical control group, the implementation of the glucose control protocol results in a significant reduction in hospital mortality (from 20.9 to 14.8%) as well as length of ICU stay decrease. There is a reduction in the incidence of new renal failure, need for red blood cell transfusion, and costs [54], but no significant difference in the occurrence of severe infections. In a prospective, randomized, controlled clinical trial [55] conducted in glucose-intolerant surgical ICU patients without diabetes, a regimen with strict insulin therapy (target glucose range, 80–120 mg/dL) induces a significant reduction in the incidence of total nosocomial infections, including the sites of intravascular devices, bloodstream, intravascular device-related bloodstream, and surgical site infections, although the therapy is associated with a higher incidence of hypoglycemia.
Discrepancies between study results can be related to several factors: differences in patient selection criteria, in statistical power and in glucose target. Therefore, the tight glycemic control reached by the intensive insulin therapy reported in the Leuven studies cannot be merely extended to all critically ill patient subgroups without bearing in mind that available literature especially for specific subgroups, is scarce and rather controversial. An accurate knowledge of previous reports and glucose metabolisms in different critical settings can help intensivists who daily face the management of glucose control in their patients.
Mechanisms explaining the acute life-saving effects of intensive insulin therapy in the ICU
Since critically ill patients suffer from hepatic and skeletal muscle insulin resistance, the mechanism by which insulin lowers blood glucose in critically ill patients is not obvious. Analyses of liver and skeletal muscle biopsies, obtained immediately after death from non-survivors in the Leuven study [16] suggest that in the critically ill patient, insulin lowers blood glucose predominantly through increased skeletal muscle glucose uptake [56]. In the critically ill patient, adipose tissue and skeletal muscle remain relatively responsive to insulin, whereas the liver is much more resistant. The β cells appear unable to compensate fully for hyperglycemia.
Other metabolic effects of insulin in the critically ill
Improvement of dyslipidemia
Insulin exerts other metabolic effects besides the control of blood glucose. Critically ill patients most characteristically show elevated triglycerides and very low levels of high density lipoprotein and low-density lipoprotein cholesterol. On the other hand, the numbers of circulating small, dense LDL particles, which presumably are more proatherogenic than the medium and large LDL particles [57], are increased [58]. Interestingly, intensive insulin therapy can in part restore this dyslipidemia, with almost complete reversal of the hypertriglyceridemia and a substantial increase in, but not normalization of the serum levels of HDL and LDL [49]. A contribution of insulin therapy to improved outcome can also be explained given the important role of triglycerides in energy provision and of lipoproteins in transportation of lipid components and endotoxin scavanging [59–61]. In the Leuven study multivariate logistic regression analysis demonstrates that the improvement of dyslipidemia with insulin therapy explains a significant part of the beneficial effect on mortality and organ failure. Surprisingly, its effects surpass those of glycemic control and insulin dose [56].
Anabolic effects
During critical illness, once a caloric deficit of 25–30 kcal/kg/d over 1 week is reached, mortality rates double [62]. Hyperglycemia per se seems to be associated with protein catabolism, as inferred by the increased rate of muscle wasting [63]. Another possible mechanism by which insulin improves outcomes is by blunting the catabolic response to stress and inflammation. In physiological conditions, the binding of insulin to its receptor suppresses proteolysis and activates protein synthesis. In animals, insulin therapy has been demonstrated to prevent weight loss [64], whereas in humans, insulin therapy has been associated with an increase in the protein content of skeletal muscle [15]. The exact mechanism explaining the anabolic effect of insulin therapy is still unknown since multiple anabolic factors, including growth hormone secretion, are affected by intensive insulin therapy [56]. It still unclear whether hyperglycemia and insulin therapy or the resolution of the underlying systemic inflammatory response is responsible for the reversal of catabolism; whatever the case, improved outcomes are noted (Fig. 2).

Metabolic derangements and systemic inflammatory response in the critically ill
Other non-metabolic effects of insulin in the critically ill
Anti-inflammatory effects
Critical illness also resembles diabetes mellitus in the activation of the inflammatory cascade, although the inflammation, as reflected by a high circulating level of C-reactive protein (CRP), in the critically ill is several times more pronounced than in diabetic patients.
Hyperglycemia per se induces a pro-inflammatory state, which includes both cellular and oxidative stress. At the cellular level, glucose is known to increase pro-inflammatory transcriptor factors (such as intranuclear NFkB binding, activator protein-1 and early growth response-1) that are suppressed by insulin. I Nevertheless, hyperglycemia can produce cellular inflammation at low levels of plasma glucose. As little as a 75-g glucose load given orally to normal subjects results in profound oxidative stress and inflammatory changes at the cellular and molecular levels [65].
According to some authors, the anti-inflammatory properties of insulin account for the beneficial effects of insulin therapy, regardless of the degree of glucose control [16, 56, 66]. In critically ill patients, intensive insulin therapy induces a significant reduction in CRP (complement reactive protein) level, as confirmed in animal models of critical illness [64]. However, it has not so far been elucidated whether insulin exerts its anti-inflammatory effects directly at the level of NFkB pathways or by lowering glucose to subsequently decrease systemic inflammation. Whatever the mechanism, the effect is rapid (observed within 2 h), and potent, since the magnitude of 2 units/h of insulin is similar to the effects of 100 mg of hydrocortisone given intravenously [67].
Preventing endothelial dysfunction and hypercoagulation
In critically ill patients [68] intensive insulin therapy reduces the circulating levels of the adhesion molecules ICAM-1 and E-selectin, independent of its effect on infection prevention. E-selectin is produced by endothelial cells, is rapidly inducible, and as such, reflects the state of the endothelium in disease. ICAM-1 is constitutively expressed at very low levels on the endothelium, and is highly inducible upon cell activation. It is also expressed by fibroblasts and hematopoietic cells. Small changes in the expression of the adhesion molecules at the endothelial cell level may go undetected in the circulation, but when differences in the amount of shedded adhesion molecules are found, this indicates a substantial change in the activity status of the endothelium. According to these authors, the lowering of ICAM-1 and E-selectin levels with intensive insulin therapy explains, at least in part, the reduced risk of developing organ failure and death with this intervention.
Anti-apoptotic effects
The literature so far available, describes only experimental models. It supports the hypothesis that insulin itself has direct cardioprotective effects during reperfusion, mainly via anti-apoptotic properties that are independent of glucose uptake [69–71]. The insulin signaling pathways involve include PI3K, Akt, and eNOS phosphorylation [69]. Whether such direct insulin-induced cell survival plays a role in mediating the observed protection of organ function in the critically ill remains unclear.
Glucose control or insulin therapy per se affects prognosis in critically ill patients
The Leuven study [16] performs a post hoc multivariate logistic regression that documents that both high insulin dosing and hyperglycemia independently predict ICU mortality, thus suggesting that the glucose-lowering effects are the key mechanism of action. Moreover, there is also a lower risk of death as blood glucose levels are lowered from 200 mg/dL to 150 mg/dL, and again to levels <110 mg/dL. Similarly in another large prospective trial [72] it is observed that control of glucose levels over absolute exogenous insulin levels accounts for the mortality improvement associated with intensive insulin therapy. These concepts are recently supported in an animal model of prolonged and critical illness (burn-injured, parentally fed rabbits) [73] The authors report that mortality is significantly lower in the two normoglycemic groups independent of insulin levels, and that maintaining normoglycemia, independent of insulin levels, prevents endothelial dysfunction as well as liver and kidney injury. Although the actions of intensive insulin therapy are clearly complex, and the exact mechanism(s) by which benefit occur is uncertain, both human and animal data strongly support glycemic control in the critical care unit by means of insulin therapy.
Conclusions
It has long been thought that hyperglycemia in the acute clinical setting may be beneficial, but recent evidence challenges this notion. In fact, several observational studies published over the last decade establish that hyperglycemia is a common abnormality in critically ill patients that is associated with increased morbidity and mortality, independent of pre-existing diabetes. Intervention studies that lower glucose with insulin demonstrate improved outcomes, suggesting that elevated glucose is a cause rather than just a marker of illness. Evidence suggests that hyperglycemia is a potentially correctable abnormality that has deleterious effects in critically ill individuals. In other words, a simple metabolic intervention, that is maintaining normoglycemia with insulin, can lead to an improvement in survival and reduced morbidity of critically ill patients.
Hyperglycemia should be regarded as a part of the systemic and complex metabolic derangements observed in critical illness in response to stress and inflammation that can lead, independent of initial disease, to multiorgan dysfunction and death. In critically ill patients, a tight glycemic control should be constantly pursued and achieved by insulin infusion remembering that the therapeutic target is fighting the systemic inflammatory response and not merely the glucose plasma levels.
References
McCowen KC, Malhotra A, Bistrian BR (2001) Stress-induced hyperglycemia. Crit Care Clin 17:107–124
Lang CH, Dobrescu C, Bagby GJ (1992) Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 130:43–52
Lind L, Lithell H (1994) Impaired glucose and lipid metabolism seen in intensive care patients is related to severity of illness and survival. Clin Intensive Care 5:100–105
Pittas AG, Siegel RD, Lau J (2004) Insulin therapy for critically ill hospitalized patients: a meta-analysis of randomized controlled trials. Arch Intern Med 164:2005–2011
Ligtenberg JJ, Meijering S, Stienstra Y, van der Horst IC, Vogelzang M, Nijsten MW, Tulleken JE, Zijlstra JG (2006) Mean glucose level is not an independent risk factor for mortality in mixed ICU patients. Intensive Care Med 32:435–438
Mizock BA (2001) Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Pract Res Clin Endocrinol Metab 15:533–551
Klip A, Tsakiridis T, Marette A, Ortiz PA (1994) Regulation of expression of glucose transporters by glucose: a review of studies in vivo and in cell cultures. FASEB J 8:43–53
Langouche L, Van den Berghe G (2006) Glucose metabolism and insulin therapy. Crit Care Clin 22:119–129
Khani S, Tayek JA (2001) Cortisol increases gluconeogenesis in humans: its role in the metabolic syndrome. Clin Sci (Lond) 101:739–747
Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M (2001) Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 534(Pt 1):269–278
Rodnick KJ, Piper RC, Slot JW, James DE (1992) Interaction of insulin and exercise on glucose transport in muscle. Diabetes Care 15:1679–1689 Review
Van den Berghe G (2004) How does blood glucose control with insulin save lives in intensive care? J Clin Invest 114:1187–1195
Quinn LA, McCumbee WD (1998) Regulation of glucose transport by angiotensin II and glucose in cultured vascular smooth muscle cells. J Cell Physiol 177:94–102
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820
Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De Wolf-Peeters C, Van den Berghe G (2005) Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 365(9453):53–59
van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R (2001) Intensive insulin therapy in critically ill patients. N Engl J Med 345:1359–1367
Van den Berghe G, Wouters PJ, Bouillon R, Weekers F, Verwaest C, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P (2003) Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Crit Care Med 31:359–366
Hirsch IB, Brownlee M (2005) Should minimal blood glucose variability become the gold standard of glycemic control? J Diabetes Complications 19:178–181
Corstjens AM, van der Horst IC, Zijlstra JG, Groeneveld AB, Zijlstra F, Tulleken JE, Ligtenberg JJ (2006) Hyperglycaemia in critically ill patients: marker or mediator of mortality? Crit Care 10:216
Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE (2002) Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 87:978–982
Cely CM, Arora P, Quartin AA, Kett DH, Schein RM (2004) Relationship of baseline glucose homeostasis to hyperglycemia during medical critical illness. Chest 126:879–887
Christiansen C, Toft P, Jørgensen HS, Andersen SK, Tønnesen E (2004) Hyperglycaemia and mortality in critically ill patients. A prospective study. Intensive Care Med 30:1685–1688
Krinsley JS (2003) Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients. Mayo Clin Proc 78:1471–1478
Whitcomb BW, Pradhan EK, Pittas AG, Roghmann MC, Perencevich EN (2005) Impact of admission hyperglycemia on hospital mortality in various intensive care unit populations. Crit Care Med 33:2772–2777
Wahab NN, Cowden EA, Pearce NJ, Gardner MJ, Merry H, Cox JL (2002) ICONS Investigators. Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era? J Am Coll Cardiol 40:1748–1754
Giannazzo G, Tola F, Vanni S, Bondi E, Pepe G, Grifoni S (2006) Prognostic indexes of septic syndrome in the emergency department. Intern Emerg Med 1:229–233
Ban KM, Rosen P (2006) Sepsis and its consequences. Intern Emerg Med 1:221–222
Krentz AJ, Natrass M (1997) Acute metabolic complications of diabetes mellitus: diabetic ketoacidosis, hyperosmolar non-ketotic syndrome and lactic acidosis. In: Pickup JC, Williams G (eds) Textbook of diabetes, 2nd edn. Blackwell, Oxford, pp 1–23
Al-Mallah M, Zuberi O, Arida M, Kim HE (2008) Positive troponin in diabetic ketoacidosis without evident acute coronary syndrome predicts adverse cardiac events. Clin Cardiol 31:67–71
Bolk J, van der Ploeg T, Cornel JH, Arnold AE, Sepers J, Umans VA (2001) Impaired glucose metabolism predicts mortality after a myocardial infarction. Int J Cardiol 79:207–214
Capes SE, Hunt D, Malmberg K, Gerstein HC (2000) Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet 355(9206):773–778
Stranders I, Diamant M, van Gelder RE, Spruijt HJ, Twisk JW, Heine RJ, Visser FC (2004) Admission blood glucose level as risk indicator of death after myocardial infarction in patients with and without diabetes mellitus. Arch Intern Med 164:982–988
Ainla T, Baburin A, Teesalu R, Rahu M (2005) The association between hyperglycaemia on admission and 180-day mortality in acute myocardial infarction patients with and without diabetes. Diabet Med 1321–1325
Aronson D, Hammerman H, Kapeliovich MR, Suleiman A, Agmon Y, Beyar R, Markiewicz W, Suleiman M (2007) Fasting glucose in acute myocardial infarction: incremental value for long-term mortality and relationship with left ventricular systolic function. Diabetes Care 30:960–966
Müdespacher D, Radovanovic D, Camenzind E, Essig M, Bertel O, Erne P, Eberli FR, Gutzwiller F (2007) On Behalf Of The Amis Plus Investigators. Admission glycaemia and outcome in patients with acute coronary syndrome. Diab Vasc Dis Res 346–52
Williams LS, Rotich J, Qi R, Fineberg N, Espay A, Bruno A, Fineberg SE, Tierney WR (2002) Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology 59:67–71
Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC (2001) Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke 32:2426–2432
Yendamuri S, Fulda GJ, Tinkoff GH (2003) Admission hyperglycemia as a prognostic indicator in trauma. J Trauma 55:33–38
Duane TM, Ivatury RR, Dechert T, Brown H, Wolfe LG, Malhotra AK, Aboutanos MB (2008) Blood glucose levels at 24 hours after trauma fails to predict outcomes. J Trauma 64:1184–1187
Wahl WL, Taddonio M, Maggio PM, Arbabi S, Hemmila MR (2008) Mean glucose values predict trauma patient mortality. J Trauma 65:42–47
Brundage SI, Kirilcuk NN, Lam JC, Spain DA, Zautke NA (2008) Insulin increases the release of proinflammatory mediators. J Trauma 65:367–372
Vogelzang M, Nijboer JM, van der Horst IC, Zijlstra F, ten Duis HJ, Nijsten MW (2006) Hyperglycemia has a stronger relation with outcome in trauma patients than in other critically ill patients. J Trauma 60:873
Van den Berghe G, Schoonheydt K, Becx P, Bruyninckx F, Wouters PJ (2005) Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology 64:1348–1353
Krinsley JS (2004) Effect of an intensive glucose management protocol on the mortality of critically ill adult patients. Mayo Clin Proc 79:992–1000
Van den Berghe G, Wilmer A, Milants I, Wouters PJ, Bouckaert B, Bruyninckx F, Bouillon R, Schetz M (2006) Intensive insulin therapy in mixed medical/surgical intensive care units: benefit versus harm. Diabetes 55:3151–3159
Malmberg K, Norhammar A, Wedel H, Ryden L (1999) Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation 99:2626–2632
Mehta SR, Yusuf S, Diaz R, Zhu J, Pais P, Xavier D, Paolasso E, Ahmed R, Xie C, Kazmi K, Tai J, Orlandini A, Pogue J, Liu L (2005) CREATE-ECLA Trial Group Investigators. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: The CREATE-ECLA randomized controlled trial. CREATE-ECLA Trial Group Investigators. JAMA 293:437–446
Malmberg K, Ryden L, Wedel H, Birkeland K, Bootsma A, Dickstein K, Efendic S, Fisher M, Hamsten A, Herlitz J, Hildebrandt P, MacLeod K, Laakso M, Torp-Pedersen C, Waldenström A (2005) DIGAMI 2 Investigators. Intense metabolic control by means of insulin in patients with diabetes mellitus and acute myocardial infarction (DIGAMI 2): effects on mortality and morbidity. Eur Heart J 26:650–661
Malmberg K, Ryden L, Hamsten A, Herlitz J, Waldenström A, Wedel H (1996) Effects of insulin treatment on cause-specific one-year mortality and morbidity in diabetic patients with acute myocardial infarction: DIGAMI Study Group. Diabetes Insulin- Glucose in Acute Myocardial Infarction. Eur Heart J 17:1337–1344
Malmberg K (1997) Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus: DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. BMJ 314:1512–1515
Malmberg K, Rydén L, Efendic S, Herlitz J, Nicol P, Waldenström A, Wedel H, Welin L (1995) Randomized trial of insulin-glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): Effects on mortality at 1 year. J Am Coll Cardiol 26:57–65
Scott JF, Robinson GM, French JM, O’Connell JE, Alberti KG, Gray CS (1999) Glucose potassium insulin infusions in the treatment of acute stroke patients with mild to moderate hyperglycemia: The Glucose Insulin in Stroke Trial (GIST). Stroke 30:793–799
Brunkhorst FM, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, Moerer O, Gruendling M, Oppert M, Grond S, Olthoff D, Jaschinski U, John S, Rossaint R, Welte T, Schaefer M, Kern P, Kuhnt E, Kiehntopf M, Hartog C, Natanson C, Loeffler M, Reinhart K (2008) German Competence Network Sepsis (SepNet). (2008) Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 358:125–139
Krinsley JS, Jones RL (2006) Cost analysis of intensive glycemic control in critically ill adult patients. Chest 129:644–650
Grey NJ, Perdrizet GA (2004) Reduction of nosocomial infections in the surgical intensive-care unit by strict glycemic control. Endocr Pract Suppl 2:46–52
Mesotten D, Swinnen J, Vanderhoydonc F, Wouters PJ, Van den Berghe G (2004) Contribution of circulating lipids to the improved outcome of critical illness by glycemic control with intensive insulin therapy. J Clin Endocrinol Metab 219–226
Kwiterovich PO (2002) Lipoprotein heterogeneity: diagnostic and therapeutic implications. Am J Cardiol 90:1i–10i
Feingold KR, Krauss RM, Pang M, Doerrler W, Jensen P, Grunfeld C (1993) The hypertriglyceridemia of acquired immunodeficiency syndrome is associated with an increased prevalence of low density lipoprotein subclass pattern B. J Clin Endocrinol Metab 1423–1427
Harris HW, Grunfeld C, Feingold KR, Read TE, Kane JP, Jones AL, Eichbaum EB, Bland GF, Rapp JH (1993) Chylomicrons alter the fate of endotoxin, decreasing tumor necrosis factor release and preventing death. J Clin Invest 1028–1034
Harris HW, Grunfeld C, Feingold KR, Rapp JH (1990) Human very low density lipoproteins and chylomicrons can protect against endotoxin-induced death in mice. J Clin Invest 696–702
Tulenko TN, Sumner AE (2002) The physiology of lipoproteins. J Nucl Cardiol 9:638–649
Bartlett RH, Dechert RE, Mault JR, Ferguson SK, Kaiser AM, Erlandson EE (1982) Measurement of metabolism in multiple organ failure. Surgery 92:771–779
Gore DC, Chinkes DL, Hart DW, Wolf SE, Herndon DN, Sanford AP (2002) Hyperglycemia exacerbates muscle protein catabolism in burn-injured patients. Crit Care Med 30:2438–2442
Weekers F, Van Herck E, Coopmans W, Michalaki M, Bowers CY, Veldhuis JD, Van den Berghe G (2002) A novel in vivo rabbit model of hypercatabolic critical illness reveals a biphasic neuroendocrine stress response. Endocrinology 143:764–774
Collier B, Dossett LA, May AK, Diaz JJ (2008) Glucose control and the inflammatory response. Nutr Clin Pract 23:3–15
Hansen TK, Thiel S, Wouters PJ, Christiansen JS, van den Berghe G (2003) Intensive insulin therapy exerts anti-inflammatory effects in critically ill patients and counteracts the adverse effect of low mannose-binding lectin levels. J Clin Endocrinol Metab 88:1082–1088
Dandona P, Thusu K, Hafeez R, Abdel-Rahman E, Chaudhuri A (1998) Effect of hydrocortisone on oxygen free radical generation by mononuclear cells. Metabolism 47:788–791
Langouche L, Vanhorebeek I, Vlasselaers D, Vander Perre S, Wouters PJ, Skogstrand K, Hansen TK, Van den Berghe G (2005) Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest 115:2277–2286
Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, Ma XL (2002) Nitric oxide mediates the anti-apoptotic effect of insulin in myocardial ischemia- reperfusion: the role of PI3-kinase, Akt, and eNOS phosphorylation. Circulation 1497–1502
Jonassen A, Sack M, Mjos O, Yellon D (2001) Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signalling. Circ Res 89:1191–1198
Jonassen A, Aasum E, Riemersma R, Mjos O, Larsen T (2000) Glucose-insulin-potassium reduces infarct size when administered during reperfusion. Cardiovasc Drugs Ther 14:615–623
Finney SJ, Zekveld C, Elia A, Evans TW (2003) Glucose control and mortality in critically ill patients. JAMA 290:2041–2047
Ellger B, Debaveye Y, Vanhorebeek I, Langouche L, Giulietti A, Van Etten E, Herijgers P, Mathieu C, Van den Berghe G (2006) Survival benefits of intensive insulin therapy in critical illness: impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes 55:1096–1105
Conflict of interest statement
The authors declare that they have no conflict of interest related to the publication of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lazzeri, C., Tarquini, R., Giunta, F. et al. Glucose dysmetabolism and prognosis in critical illness. Intern Emerg Med 4, 147–156 (2009). https://doi.org/10.1007/s11739-008-0206-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-008-0206-3