
Abstract
Drought stress inhibits rice growth and biomass accumulation. To identify novel regulators of drought-stress responses in rice, we conducted a proteome-level study of the stress-susceptible (SS) Oryza sativa L. cv. ‘Leung Pratew 123’ and its stress-resistant (SR) somaclonal mutant line. In response to osmotic-stress treatments, 117 proteins were differentially accumulated, with 62 and 49 of these proteins detected in the SS and SR rice lines, respectively. There were six proteins that accumulated in both lines. The proteins in the SS line were mainly related to metabolic processes, whereas the proteins identified in the SR line were primarily related to retrotransposons. These observations suggest that retrotransposons may influence the epigenetic regulation of gene expression in response to osmotic stress. To identify the biological processes associated with drought tolerance in rice, we conducted a co-expression network analysis of 55 proteins that were differentially accumulated in the SR line under osmotic-stress conditions. We identified a major hub gene; LOC_Os04g38600 (encoding a glyceraldehyde-3-phosphate dehydrogenase), suggesting that photosynthetic adaptation via NADP(H) homeostasis contributes to drought tolerance in rice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among constraining environmental conditions, drought is the most crucial factor that limits plant growth, development, and productivity (Boyer 1982). Osmotic stress induces a series of molecular and physiological responses, including stomatal closure, osmotic adjustment, reduced photosynthetic activity, and abscisic acid (ABA) synthesis. Decreased water loss due to stomatal closure and/or reduced stomatal density is an example of a drought-avoidance mechanism (Chaves et al. 2003; Hadiarto and Tran 2010). However, a consequence of this strategy is often reduced photosynthesis and growth because of decreased carbon uptake.
Under stressful conditions, the earliest plant responses generally involve changes to gene expression (Yamaguchi-Shinozaki and Shinozaki 2006). These stress-induced changes in gene expression can alter plant protein profiles. However, there is not always a strong correlation between gene expression and protein abundance, often because of post-translational modifications (Park 2004; Urano et al. 2010; Webster and Thomas 2012). Therefore, to clarify plant responses to environmental stimuli, a proteome-level investigation may be a better option for revealing cellular adaptations (Kosová et al. 2015).
Proteomics-based studies have been used to identify drought-responsive proteins and genes in many crops, including rice (Ali and Komatsu 2006; Chamnanmanoontham et al. 2015; Ji et al. 2012), cotton (Deeba et al. 2012), grapevine (Lovisolo et al. 2010), soybean (Deshmukh et al. 2014; Oh and Komatsu 2015), wheat (Alvarez et al. 2014; Ford et al. 2011), and watermelon (Akashi et al. 2011). Different proteomics techniques, including two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) with subsequent mass spectrophotometry identification (Ali and Komatsu 2006; Ji et al. 2012) or gel-free approaches (Oh and Komatsu 2015), have been used to identify important genes involved in abiotic stress responses. We herein describe the use of gel-based liquid chromatography–tandem mass spectrometry (GeLC–MS/MS) to overcome the limitations of 2D-PAGE, which is believed to be ill-suited for analyzing very high- and low-molecular-weight proteins, as well as hydrophobic, highly acidic, or alkaline proteins (Santoni et al. 2000). The accessibility of low-abundant proteins can be enhanced using sodium dodecyl sulfate (SDS)-PAGE/nano-LC–MS/MS rather than 2D-PAGE (Abere et al. 2012; Tefon et al. 2011; Wolff et al. 2006).
The increase of bioinformatics tools, which can be used for finding a novel gene or studying gene ontology, creates the extensive use of omics data. A gene co-expression network is one of the tools which can imply the complex gene interaction and provide us the understanding of plant process in particular hypothesis (Aoki et al. 2007; Usadel et al. 2009). The complexity of biological networks can classify into levels of genome-wide organization and individual molecular components such as node, degree, and module. Module is a word used to represent a group of highly co-express genes associated with biological processes which cannot be attributed to a single gene (Hartwell et al. 1999). A co-expression network has uncovered novel stress-responsive gene in many organisms such as rice (Chamnanmanoontham et al. 2015; Nounjan et al. 2016; Smita et al. 2015) Escherichia coli and yeast (Altaf-Ul-Amin et al. 2006).
Rice (Oryza sativa L. spp. indica) is staple food for more than one-third of the global population. However, rice production is limited by environmental stresses such as cold (Jia et al. 2015), drought (Haefele et al. 2016; Izanloo et al. 2008; Saikumar et al. 2016), and salinity (Javid et al. 2011). In this study, we conducted a proteome-level investigation of two Thai rice lines with contrasting stress tolerances. We used the stress-susceptible ‘Leung Pratew 123’ cultivar (LPT123; SS) and the stress-tolerant somaclonal line derived from LPT123 (i.e., LPT123–TC171; SR). The SR line exhibits greater tolerance to osmotic (Pongprayoon et al. 2013; Thikart et al. 2005) and salt (Thikart et al. 2005; Udomchalothorn et al. 2009) stresses. Some of the genes responsible for the increased salt-stress tolerance have been identified (Pongprayoon et al. 2013; Sripinyowanich et al. 2013; Thikart et al. 2005; Udomchalothorn et al. 2009, 2014; Vajrabhaya and Vajrabhaya 1991). To evaluate the potential mechanisms regulating osmotic-stress tolerance, we completed a GeLC–MS/MS proteome-level analysis of the SR line. A co-expression network, constructed based on the proteins that were significantly over- or under-accumulated in the SR line in response to osmotic stress, was analyzed to identify the hub gene(s), which are the genes with an extremely high connectivity to other genes in the module showing the high degree of co-expression with other genes, suggesting the involvement in osmotic adaptation. Differential transcription of these genes was confirmed, and their potential role in rice drought tolerance is discussed. This proteome-level investigation of closely related rice lines differing in osmotic-stress tolerance (Thikart et al. 2005) revealed a novel group of proteins and a mechanism regulating osmotic-stress adaptation in rice.
Materials and methods
Proteomic analysis of two rice lines
Plant materials and growth conditions
The SS and SR rice lines used in all experiments have been previously described (Chamnanmanoontham et al. 2015; Pongprayoon et al. 2013; Sripinyowanich et al. 2013; Thikart et al. 2005; Udomchalothorn et al. 2009, 2014; Vajrabhaya and Vajrabhaya 1991). The SS and SR lines were grown using a completely randomized design with three biological replicates. Rice germination and growth conditions were similar to those described, previously (Chamnanmanoontham et al. 2015). Briefly, rice seeds were soaked in distilled water for 24 h and then germinated on sterilized sand flooded with distilled water under natural light. Two weeks after germination, modified WP solution (Vajrabhaya and Vajrabhaya 1991) was added and seedlings were grown in the greenhouse under natural light, which has the intensity of 93–99 μmol photon m−2 s−1 and a relative humidity of between 74 and 81%. The nutrient solution was refreshed every 7 days. After 4 weeks, seedlings of each line were separated into two groups. The plants of one group continued to grow in freshly prepared nutrient solutions, while plants of the other group were transferred to nutrient solution supplemented with 10% (w/v) polyethylene glycol 6000 (PEG6000) to simulate osmotic-stress conditions (Pongprayoon et al. 2013). We collected SS and SR leaves at 0, 2, 6, and 24 h after treatment, then, immediately froze them in liquid nitrogen and kept at −80 °C until analyzed. The proteins from SS and SR at each timepoint of collection were extracted separately for proteomics analysis.
Protein extraction and one-dimensional SDS-PAGE
Leaf tissue from three seedlings was pooled for each replicate, and proteins were extracted from ground tissue by incubating samples in 0.1% SDS at 37 °C for 3 h. Total protein concentrations were determined using an established procedure (Lowry et al. 1951), with bovine serum albumin serving as the protein standard. Equal amounts of extracted proteins were separated on a one-dimensional 12.5% (w/v) SDS-PAGE gel (Laemmli 1970), which was stained with Coomassie Blue R-250 to visualize proteins (Meyer and Lamberts 1965).
Trypsin digestion for mass spectrometry analysis
Proteins were manually excised from the SDS-PAGE gel using a previously described method (Jaresitthikunchai et al. 2009). The in-gel digestion procedure was conducted at the Proteomics Laboratory, National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, Thailand. Gel lanes were divided into six groups according to molecular weight (with protein markers as standards) to reduce the complexity of the peptide analysis. The gel was cut into 1 mm3 pieces that were individually washed with sterile milli-Q water. The gel pieces were then dehydrated with 100% acetonitrile (ACN) for 5 min and then dried for 5 min. Disulfide bonds were reduced by incubating gel pieces in a solution consisting of 10 mM dithiothreitol and 10 mM NH4HCO3 for 1 h. The gel pieces were then alkylated with 100 mM iodoacetamide and 10 mM NH4HCO3 for 1 h in the dark. Subsequently, the gel pieces were dehydrated three times for 5 min each in 100% ACN. Proteins were digested in a trypsin solution (10 ng µl−1 trypsin, 50% ACN, and 10 mM NH4HCO3) for 20 min and then incubated in 30% ACN overnight. The digested peptides were treated with a solution consisting of 50% ACN and 0.1% trifluoroacetic acid for 10 min. All procedures were completed at room temperature, except for the drying of extracted peptides, which occurred in an oven at 40 °C. Dried peptides were stored at −80 °C prior to MS analyses.
Liquid chromatography–electrospray ionization tandem mass spectrometry
The digested peptide solutions were injected into an Ultimate 3000 LC system (Dionex, CA, USA) equipped with an ESI-Ion Trap mass spectrometer (HCT Ultra PTM Discovery System, Bruker Daltonik GmbH, Germany). The electrospray flow rate to the µ-precolumn (Monolithic Trap Column; 200 µm internal diameter × 5 cm) was set at 20 µl min−1. Peptides were separated on a nano column (Monolithic Column; 100 µm internal diameter × 5 cm) at a mobile phase flow rate of 1.0 µl min−1. Three technical replications were performed.
The raw LC–MS/MS data were converted to mzXML format using the CompassXport 1.3.10 program (Bruker Daltonik GmbH, Germany). We used the DeCyder MS Differential Analysis software [GE Healthcare (Johansson et al. 2006; Thorsell et al. 2007)] to quantify proteins. The analyzed MS/MS data were checked against the National Center for Biotechnology Information non-redundant database 20170221 using Mascot software [Matrix Science, London, UK (Perkins et al. 1999)]. We used the following parameters: taxonomy: O. sativa (rice); enzyme: trypsin; allow up to: 1 missed cleavage; fixed modifications: carbamidomethyl; variable modifications: oxidation; peptide tolerance: ±1.2 Da; MS/MS tolerance: ±0.6 Da; peptide charge stage: 1+, 2+, and 3+ (monoisotopic); and instrument: ESI-TRAP.
Identification of genes/proteins and analysis of a co-expression network
The identified proteins were searched against the Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu) (Kawahara et al. 2013) using BLASTP to annotate proteins and assign functions based on gene ontology. If more than one locus was predicted, the highest Mascot score or the lowest ANOVA P value (if the Mascot scores were the same) was used to determine the most likely locus according to the DeCyder MS Differential Analysis software. The identified proteins in each set of treatments that matched the above criteria were visualized and analyzed with the MultiExperiment Viewer (MeV) program to identify the proteins with significantly differential expression between unstressed plants and osmotic-stress-treated plants at different timepoints with t test (P < 0.05) (Saeed et al. 2003). The hierarchical clustering was conducted using the Pearson correlation coefficient. The co-expression network analysis of proteins that were significantly affected by osmotic stress in the SR line was generated using a ‘guide gene approach’ by RiceFREND with hierarchy of 2 and mutual rank (MR) of 5 (Sato et al. 2013).
Detection of osmotic-stress-responsive genes in rice
Shoots of 7-day-old SS and SR rice seedlings were cut and air-dried for 2 h to simulate drought-stress conditions, similar to the method used by (Huang et al. 2010, 2011) used three replicates for each set. Total RNA was extracted with PureLink Plant RNA Reagent (Invitrogen, USA), treated with DNase I (Thermo Fisher Scientific, USA) to cleave genomic DNA, and purified by phenol–chloroform precipitation. The purified RNA (1 µg) was reverse-transcribed to generate first-strand cDNA using the iScript Reverse Transcription Supermix (Bio-Rad, USA). The major hub gene from the co-expression network, LOC_Os04g38600, was selected for transcriptional-level analysis. A semi-quantitative reverse transcription (RT) polymerase chain reaction (PCR) experiment was conducted in 50-μl samples containing Taq DNA Polymerase (Thermo Fisher Scientific). Primers specific for the selected genes (Supplementary Table 1) were designed based on coding sequences, which were retrieved from the Rice Genome Annotation Project database (Kawahara et al. 2013). DREB2A (Dubouzet et al. 2003) and OsEF-1α (Saeng-ngam et al. 2012) were used as the stress-responsive gene and internal controls, respectively.
Results and discussion
Drought stress induced by 10% PEG6000 treatment altered rice leaf protein profiles
Leaf proteins from unstressed and osmotic-stress-treated SS and SR rice lines were analyzed by GeLC–MS/MS. A total of 357 proteins (Supplementary Table 2) were detected in the SS and SR lines. Based on the comparison of proteins by the MeV program, 68 and 55 proteins from the SS and SR lines, respectively, were significantly up- or down-regulated in stressed plants relative to their levels in untreated control plants (P ≤ 0.05) (Fig. 1a, b). It is worth mentioning that more than 1200 proteins were identified by the MascotTM software. Some had very short peptide sequences, containing only five amino acid residues. After the using Blastp in NCBI database (Coordinators 2016), only 357 proteins showed the highest similarity with rice proteins. Some of the proteins identified were not annotated by either MSU Rice Genome Annotation Project (Kawahara et al. 2013) or International Rice Genome Sequencing Project (Kawahara et al. 2013; Sakai et al. 2013). Therefore, the locus numbers of these proteins were not listed in the Supplementary Table 2. These data reveal the difference in annotation among various databases.

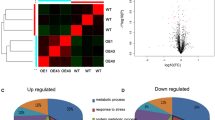
Heat map of significantly up- or down-regulated leaf proteins in 4-week-old SS (a) and SR (b) rice plants at 0, 2, 6, and 24 h after osmotic-stress treatment with 10% PEG6000. The heat map was created using the MultiExperiment Viewer program. Columns represent treatments and harvest time after treatment. Rows correspond to individual proteins that were significantly up- or down-regulated by osmotic-stress treatment, relative to the levels in control plants. Light green-to-dark red bars indicate low-to-high protein abundances. Venn diagram of significant proteins (c). There were 62 (group a) and 49 (group b) identified proteins present only in the SS or SR lines, respectively. Six (group c) identified proteins were detected in both lines. The identified proteins are listed in Supplementary Tables 2 and 3
The SS and SR proteins were categorized into ten functional groups (Fig. 2; Supplementary Table 3). The number of proteins associated with metabolic process, defense, signaling, cellular process, transposon, and post-translation was higher in SS plants than in SR plants. Proteins associated with retrotransposon, transcription, post-transcription, and transport were more abundant in the SR rice line, suggesting the importance of changes in these functions for drought tolerance (Fig. 2).

Functional classification of drought-responsive proteins detected in LPT123 (SS) (a) and LPT123–TC171 (SR) (b) rice leaves. The functions were categorized according to gene ontology annotations from the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu)
Disregarding the proteins with unknown functions, retrotransposons were the main group of proteins affected by osmotic stress in SR plants, while proteins related to metabolic processes were the most commonly affected proteins in SS plants. The categories of post-translation were the group found only in SS line, while post-transcription was the category found only in SR plants. These differences suggest that SS and SR respond differently to osmotic stress.
The SR line was generated because of somaclonal variations in the SS line, and selected under salt-stress conditions for six generations (Sripinyowanich et al. 2013; Vajrabhaya and Vajrabhaya 1991). A large proportion of mutations in rice cell cultures are caused by the transposition of retroelements (Hirochika et al. 1996). Thus, the SR line may carry conserved retrotransposon genes within its genome. Retrotransposons are activated by multiple stresses (Alzohairy et al. 2012), including drought (Kapazoglou et al. 2013). The mechanism responsible for this activation under drought conditions is hypomethylation (Wang et al. 2011). In addition, drought stress induces epigenetic changes through DNA methylations, with 70% of methylated DNA being demethylated after stressed plants are returned to normal growth conditions (Wang et al. 2011). Grandbastien (2015) reviewed the up-regulation of the long terminal repeat (LTR) retrotransposons under various stress conditions. The ‘EG4’ rice cultivar contains more than 1000 mPing miniature inverted-repeat transposable elements (Naito et al. 2009). The expression of many genes containing the mPing element can be induced by exposure to salt or cold stress (Lisch 2013). In perennial ryegrass, drought stress leads to enhanced DNA demethylation and up-regulated expression of demethylated genes (Tang et al. 2014). The drought-tolerant Macrotyloma uniflorum (Lam.) Verdc. genotype exhibits lower methylation levels than the drought-susceptible genotype (Bhardwaj et al. 2013). The demethylation of retroelements under drought-stress conditions is correlated with the expression of nearby genes, which may represent the mechanism regulating stress-induced or repressed gene expression.
Drought-responsive genes in the SS and SR lines
According to osmotic-stress-induced differential expression of 68 proteins in SS line and 55 proteins in SR line, 6 of them were significantly affected by osmotic stress in both rice lines (Fig. 1c). The proteins up-regulated in both rice lines in response to osmotic stress included a helicase domain-containing protein, cytochrome P450, and stripe rust-resistance protein Yr10 (Table 1). The families of these three proteins are known to be involved in stress responses (Baldoni et al. 2015; Barak et al. 2014; Gao et al. 2014; Himmelbach et al. 2002; Kant et al. 2007).
The other three proteins (i.e., LOC_Os01g56200, LOC_Os04g35864, and LOC_Os07g29820) were affected differently in drought-stressed SS and SR lines. All of them were up-regulated in the SR line, but down-regulated in the SS line.
RNA helicase activity involves an ATP-driven unwinding of an RNA duplex, and has been observed in several organisms such as viruses, bacteria, humans, and plants. It plays an important role during cell growth and in responses to abiotic stresses. The DEAD-box genes, including STRS1, STRS2, TaRH1, SlDEAD31, and OsBAT1, form the largest group of RNA helicase genes (Barak et al. 2014; Chen et al. 2014; Kant et al. 2007; Zhang et al. 2014; Zhu et al. 2015). In Triticum aestivum, low temperature, dehydration, and salt stresses lead to an accumulation of an RNA helicase (TaRH1) (Zhang et al. 2014). In tomato, SlDEAD31 expression is promoted by heat, cold, and dehydration stresses, and SlDEAD31 overexpression results in increased salt tolerance and drought resistance (Zhu et al. 2015). Transgenic rice overexpressing OsBAT1 can germinate and grow on Murashige and Skoog medium supplemented with 200 mM NaCl (Tuteja et al. 2015). OsSUV3, encoding DNA/RNA helicase and belonging to the Ski2 family of DExH/D-box helicases, was shown to function in salt tolerance in rice by maintaining photosynthesis and antioxidant machinery (Tuteja et al. 2014). Therefore, the helicase domain-containing protein detected in this study may be active in rice drought-stress responses.
A cytochrome P450, CYP707A family member was identified as ABA 8′-hydroxylase, which degraded ABA under dehydration stress condition. The knockout mutant of CYP707A3 gene led to drought-tolerant phenotype (Umezawa et al. 2006). However, the ectopic expression of PtCYP714A3 from Populus trichocarpa improved salt tolerance in transgenic rice (Wang et al. 2016). Moreover, the expression of Os08g01480, encoding CYP-like protein, in Arabidopsis, caused the tolerance to heavy metal, salt, and dehydration stress (Rai et al. 2015). The up-regulated cytochrome P450 (LOC_Os10g05020) suggests the involvement of this protein in osmotic-stress response.
Stripe rust-resistance protein is encoded from Yr10, which is one of the disease-resistant (R) genes in plants. It has evolutionary-conserved and unique CC–NBS–LRR sequence (Liu et al. 2014). This gene is conserved among plant species, including wheat, maize, sorghum, and rice. Another NBS–LRR disease-resistance protein, LOC_Os07g29820, was also found to be up-regulated in SR line, but down-regulated in SS line, which was similar to the expression of another defense protein, LOC_Os01g56200. These proteins may have the function not only in biotic stress responses, but also in abiotic responses.
DDT domain has been characterized as a domain in bromodomain PHD finger transcription factors (BPTFs) (Doerks et al. 2001). It was shown to have the DNA-binding function. However, the function of DDT domain-containing protein encoded from LOC_Os04g35864 has not been reported.
Specific proteins detected only in the SR line include transposable elements, and proteins involved in metabolic processes and signaling
Transposable elements
The genes encoding the proteins that accumulated only in the drought-tolerant line in response to osmotic stress may be useful as drought-tolerance genes. Protection from environmental stresses may be mediated by epigenetic events, such as the induction of the expression of adjacent genes by transposable elements. More than one-fifth of 49 proteins detected only in the SR line (Supplementary Table 4) consisted of a combination of retrotransposons and transposons. Transposable elements (TEs) are classified as Class I (copy-and-paste mechanism via an RNA intermediate or retroelement) or Class II (cut-and-paste mechanism via a DNA intermediate) transposons, and are major components of eukaryotic genomes (Anca et al. 2014; Chadha and Sharma 2014). In addition, the LTR retrotransposons, which may mediate somaclonal variation, are the major plant TEs (Grandbastien 2015; Wessler 1996). For example, copper and heat shock stresses induce TE activities, leading to instability in the Magnaporthe oryzae genome (Chadha and Sharma 2014). The Hordeum vulgare DEMETER gene (HvDME) contains an LTR retrotransposon element. Its expression is induced in drought-tolerant barley exposed to drought conditions, resulting in differential DNA methylation in drought-sensitive (e.g., ‘Caresse’) and drought-tolerant (e.g., ‘Demetra’) cultivars (Kapazoglou et al. 2013). The activation of TEs is one of the mechanisms that enables self-protection and self-repair. It also stimulates the expression of other genes responsible for stress responses (Grandbastien 2015).
Plant metabolism
Several proteins involved in metabolic processes were up- or down-regulated following treatment with osmotic stress (Supplementary Table 4). When plant cells experience abiotic stress, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is one of the most prominent proteins targeted for oxidative modification (Hildebrandt et al. 2015). This enzyme has an important role in converting glycerate-3-phosphate to glyceraldehyde-3-phosphate. Glycerate-3-phosphate is an electron acceptor that receives electrons from NADPH and protects photosystem II from reactive oxygen species (Takahashi and Murata 2006). In our study, GAPDH (LOC_Os04g38600) abundance increased in only SR lines treated with osmotic stress for 2 h, implying that the enzyme has a role in protecting photosystem II. This is similar to the findings for two wheat cultivars with contrasting drought tolerances. The GAPDH enzyme is up-regulated after 48 h of PEG6000 treatment in both wheat genotypes (Cheng et al. 2015). However, in our study, the maintenance of stable GAPDH levels for up to 24 h of osmotic stress was observed only in the SR line (Fig. 3B), suggesting that photosystem II is protected from the effects of osmotic stress more in the SR line than in the SS line. A proteome-level study of Thellungiella halophila chloroplasts exposed to different saline conditions uncovered several salt-responsive proteins, including the GAPDH β subunit (GAPB) (Chang et al. 2015). Overexpression of GAPB in transgenic A. thaliana increases the chlorophyll concentration, dry weight, water content, and survival rate.

Co-expression networks of the significant changed proteins from SR line. A–G Genes significantly expressed in SR lines. Squares represent the transcription factors. Blue circles indicate nodes in the network, while the green, red, and pink circles in the ellipses represent the metabolic pathways in which the node genes (ellipses) are involved
Enolase, the enzyme in glycolytic pathway, was the protein involving in metabolic process with significant reduction found only in SR line. In a previous study, enolase protein abundance was significantly higher in drought-tolerant Chinese spring wheat than in a drought-sensitive cultivar after a 48-h PEG6000 treatment (Cheng et al. 2015). This pointed out that drought tolerance in different species might use different metabolic pathways for adaptation.
Both GAPDH and enolase changes suggested the adaptation in carbohydrate metabolism to drought stress in SR line. The regulation of photosynthetic efficiency under drought stress leads to the maintenance of grain yield in rice (Ambavaram et al. 2014). Sugar accumulation is also the mechanisms for tolerance to abiotic stresses, including drought (Pandey and Shukla 2015), salt (Udomchalothorn et al. 2009), and chilling stresses (Morsy et al. 2007).
Plant signaling and defense
Osmotic stress caused the changes in proteins with signaling and defense functions differently between SS and SR lines. These suggested that these two lines had different signaling pathways and use different defense responses to cope with osmotic stress.
Other mechanisms
Transcription and post-transcriptional regulation for osmotic-stress response have been investigated in various plant species. In this study, at least two transcription factors, Trihelix transcription factor GTL1 and WKRY106 were found to have a change in protein levels in SR line, but not in SS line, suggesting the role in the regulation of osmotic-stress tolerance. Further validation is required for the future study. The reduction of GTL1 in SR line is consistent with the previous study in Arabidopsis, showing that A. thaliana GT-2 LIKE 1 (AtGTL1) loss-of-function mutant (gtl1-4) exhibited a higher integrated water use efficiency and a higher survival rate after exposure to water deficit conditions (Yoo et al. 2010).
Post-transcriptional regulation of gene expression is controlled by gene activities in mitochondria. In this experiment, pentatricopeptide repeat (PPR) protein was up-regulated after drought stress. Mitochondrial pentatricopeptide repeat (PPR) proteins are associated with many plant biological processes, including RNA sequence changes, translation, and seed and embryo development. Salt, ABA, and oxidative stresses inhibit plant growth in an A. thaliana mutant (ppr40), and results in the accumulation of reactive oxygen species. Because PPR proteins are very important to plant organelles, defects in these proteins lead to retarded growth, diverse defects in embryo morphology, and irregular photosynthesis (Cushing et al. 2005; Manna 2015; Meierhoff et al. 2003; Pusnik et al. 2007).
We identified one protein with transport functions (i.e., SEC 14 cytosolic factor family protein). A comparison of transcriptomes among several sorghum genotypes revealed that SEC14 cytosolic factor protein is more abundant in the nitrogen stress-tolerant sorghum genotypes than in the susceptible sorghum lines. In addition, the production of this protein can lead to greater membrane stability and stress tolerance (Gelli et al. 2014).
Co-expression network reveals GAPDH involving photosynthetic adaptation during drought stress
A group of 57 genes identified only in the SR line was subjected to co-expression network analysis using the RiceFREND (Sato et al. 2013). Seven proteins showed the co-expression network, as shown in Fig. 3. The seven proteins with the co-expressed gene network were transcription factor GTL1 (Fig. 3A), cytochrome P450 (LOC_Os12g04100) (B), GAPDH (C), LOC_Os08g17020 (expressed protein, D), Tubulin/ftsz domain-containing protein (E), cytochrome P450 (LOC_Os10g05020) (F), and stripe rust-resistance protein Yr10 (G). Nodes F and G were also expressed in SS line, while nodes A–E were the proteins significantly changed only in SR line.
Among significant proteins expressing only in SR line, with co-expressed gene network, GAPDH was the only protein showing the connection to the genes in metabolic pathways, especially the genes in photosynthesis. This suggested the importance of the GAPDH in osmotic-stress response. We conducted semi-quantitative RT-PCR to validate the expression of GAPDH genes. We used EF1-α as an internal control, while DREB2A served as a stress-responsive gene control. Although GAPDH expression was up-regulated in both SS and SR leaves treated with drought conditions for 2 h, the increase was greater in the SR leaves (Fig. 4). This was consistent with the increase in GAPDH protein abundance during our proteome-level analysis (Fig. 5). These observations indicate that these genes are regulated at the transcriptional level.

Semi-quantitative expression of GAPDH and FNR in SS and SR leaves after air-dry treatment

Protein expression patterns for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) after an osmotic-stress (10% PEG6000) treatment for 0, 2, 6, and 24 h
In many species, the up-regulation of GAPDH is associated with drought tolerance. For example, the drought-tolerant ‘Ningchun 47’ wheat cultivar increases GAPDH accumulation under drought conditions (Cheng et al. 2015), while the GAPB expression level increases in T. halophila under salt-stress conditions. A previous study revealed that GAPDH plays an important role in maintaining photosynthetic activities and plant development under salt-stress conditions (Chang et al. 2015). The overexpression of a potato GAPDH gene in transgenic tobacco enhances drought tolerance, while reduced expression of this gene leads to a severe stress phenotype under drought-stress conditions, suggesting the importance of GAPDH in drought tolerance (Kappachery et al. 2014).
Ferredoxin–NADP reductase (FNR) is important for balancing electron transport and redox homeostasis in chloroplasts. Therefore, the gene expression of FNR was investigated in both lines, as shown in Fig. 4. The decrease of FNR expression was detected only in SR line after stress. The abundance of this enzyme decreases after drought-stress treatments in transgenic tobacco (Gharechahi et al. 2015), P. cathayana (Xiao et al. 2009), wheat (Budak et al. 2013), and rice (Nouri et al. 2015), which is consistent with our results (Fig. 4). In contrast, the expression of ferredoxin genes significantly increases in potato after an incubation at a moderately high temperature (30 °C) (Hancock et al. 2014). In addition, maize FNR levels increase after being treated with 25 mM NaCl (Zörb et al. 2009) or 16% PEG (Tai et al. 2011). However, in some wheat cultivars, FNR exhibits no significant changes when grown under drought-stress conditions (Nikolaeva et al. 2010). These findings imply that different species use different mechanisms to balance electron flow in photosynthetic processes under osmotic-stress conditions.
Both GAPDH and FNR are involved in regulating plant NADP(H) levels (Hald et al. 2008). Drought stress induces a decrease in stomatal conductance, leading to lower Calvin cycle activities, and ultimately an increase in stromal NADPH levels. In addition, the repair of photosystem II from photodamage is suspended by the decreasing of Calvin cycle activities and the disruption of Calvin cycle diminish their utilization of NADPH which induces ROS accumulation (Takahashi and Murata 2005; Zavafer et al. 2015). Therefore, maintaining an NADP(H) homeostasis is needed for preventing PSII damage. In addition, reduced FNR levels under drought conditions also contribute to NADPH homeostasis, delaying a NADPH production, whereas increased GAPDH level will decrease NADPH/NADP ratios which lead to protection of PSII. Consistent with our result, SR line shows a greater reduction after the stress, whereas FNR slightly down-regulated after dehydration (Fig. 4). In drought-tolerant transgenic tobacco plants producing cyanobacterial flavodoxin, the abundance of FNR decreases after exposure to drought stress (Gharechahi et al. 2015). In conclusion, SR rice showed that GAPDH were up-regulated, while FNR reduced under the stress (Fig. 4) which imply that during the stress, plants try to use NADP(H) homeostasis mechanism to prevent photosystem damage by stress.
Conclusion
Our proteome-level analysis revealed several candidate proteins with important roles in drought responses. We determined that the genes encoding GAPDH and FNR, which are key enzymes influencing NADP(H) homeostasis, are affected by osmotic-stress treatments. These findings suggest that drought tolerance in rice may be mediated by photosynthesis-related adaptations involving NADP(H) homeostasis. Our research confirms the value of conducting proteome-level investigations to further characterize the potential mechanism regulating drought tolerance.
Author contribution statement
Six authors contribute to this manuscript as follows: study conception and design: SC and NC. Acquisition of data: NC, MM, NP, and SR. Technical support: NP and SR. Analysis and interpretation of data: NC and SC. Drafting of manuscript: NC and SC. Critical revision: MM and SC. Final approval of the article: SC.
Abbreviations
- AKT1:
-
Inward-rectifier K+ channel
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GAPB:
-
GAPDH β subunit
- GeLC–MS/MS:
-
Gel-based liquid chromatography–tandem mass spectrometry
- GORK:
-
Guard cell ‘outward-rectifying’ K+ channel
- NBS-LRR:
-
Nucleotide-binding site–leucine-rich repeat
- OSBPs:
-
Oxysterol-binding proteins
- PEG6000:
-
Polyethylene glycol 6000
- PP2C:
-
Protein phosphatase 2C
- PPR:
-
Pentatricopeptide repeat
- SKOR:
-
Stelar K+ ‘outward-rectifying’ channel
- SR:
-
Stress resistant
- SS:
-
Stress susceptible
- TE:
-
Transposable element
References
Abere B et al (2012) Proteomic analysis of Chikungunya virus infected microgial cells. PLoS One 7:e34800. doi:10.1371/journal.pone.0034800
Akashi K et al (2011) Dynamic changes in the leaf proteome of a C-3 xerophyte, Citrullus lanatus (wild watermelon), in response to water deficit. Planta 233:947–960
Naito K et al (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461:1130–1134. http://www.nature.com/nature/journal/v461/n7267/suppinfo/nature08479_S1.html
Ambavaram MMR et al (2014) Coordinated regulation of photosynthesis in rice increases yield and tolerance to environmental stress. Nat Commun 5:5302. http://www.nature.com/articles/ncomms6302#supplementary-information
Ali GM, Komatsu S (2006) Proteomic analysis of rice leaf sheath during drought stress. J Proteome Res 5:396–403
Altaf-Ul-Amin M, Shinbo Y, Mihara K, Kurokawa K, Kanaya S (2006) Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinform 7:207. doi:10.1186/1471-2105-7-207
Alvarez S, Choudhury SR, Pandey S (2014) Comparative quantitative proteomics analysis of the ABA response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J Proteome Res 13:1688–1701
Alzohairy A, Yousef M, Edris S, Kerti B, Gyulai G, Bahieldin A (2012) Detection of LTR retrotransposons reactivation induced by in vitro environmental stresses in barley (Hordeum vulgare) via RT-qPCR. Life Sci J 9:5019–5026
Anca I-A, Fromentin J, Bui QT, Mhiri C, Grandbastien M-A, Simon-Plas F (2014) Different tobacco retrotransposons are specifically modulated by the elicitor cryptogein and reactive oxygen species. J Plant Physiol 171:1533–1540. doi:10.1016/j.jplph.2014.07.003
Aoki K, Ogata Y, Shibata D (2007) Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol 48:381–390. doi:10.1093/pcp/pcm013
Baldoni E, Genga A, Cominelli E (2015) Plant MYB transcription factors: their role in drought response mechanisms. Int J Mol Sci 16:15811–15851. doi:10.3390/ijms160715811
Barak S, Yadav NS, Khan A (2014) DEAD-Box RNA helicases and epigenetic control of abiotic stress-responsive gene expression. Plant Signal Behav 9:e977729–e977734. doi:10.4161/15592324.2014.977729
Bhardwaj J, Mahajan M, Yadav SK (2013) Comparative analysis of DNA methylation polymorphism in drought sensitive (HPKC2) and Tolerant (HPK4) genotypes of horse gram (Macrotyloma uniflorum). Biochem Genet 51:493–502. doi:10.1007/s10528-013-9580-2
Boyer JS (1982) Plant productivity and environment. Science 218:443–448
Budak H, Akpinar BA, Unver T, Turktas M (2013) Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanoLC–ESI–MS/MS. Plant Mol Biol 83:89–103. doi:10.1007/s11103-013-0024-5
Chadha S, Sharma M (2014) Transposable elements as stress adaptive capacitors induce genomic instability in fungal pathogen Magnaporthe oryzae. PLoS One 9:e94415
Chamnanmanoontham N, Pongprayoon W, Pichayangkura R, Roytrakul S, Chadchawan S (2015) Chitosan enhances rice seedling growth via gene expression network between nucleus and chloroplast. Plant Growth Regul 75:101–114. doi:10.1007/s10725-014-9935-7
Chang L et al (2015) The beta subunit of glyceraldehyde 3-phosphate dehydrogenase is an important factor for maintaining photosynthesis and plant development under salt stress—based on an integrative analysis of the structural, physiological and proteomic changes in chloroplasts in Thellungiella halophila. Plant Sci 236:223–238. doi:10.1016/j.plantsci.2015.04.010
Chaves MM, Maroco JP, Pereira JS (2003) Understanding plant responses to drought—from genes to the whole plant. Funct Plant Biol 30:239–264
Chen J, Zhang Y, Liu J, Xia M, Wang W, Shen F (2014) Genome-wide analysis of the RNA helicase gene family in Gossypium raimondii. Int J Mol Sci 15:4635–4656. doi:10.3390/ijms15034635
Cheng Z et al (2015) Identification of leaf proteins differentially accumulated between wheat cultivars distinct in their levels of drought tolerance. PLoS One 10:e0125302. doi:10.1371/journal.pone.0125302
Coordinators NR (2016) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 44:D7–D19. doi:10.1093/nar/gkv1290
Cushing DA, Forsthoefel NR, Gestaut DR, Vernon DM (2005) Arabidopsis emb175 and other ppr knockout mutants reveal essential roles for pentatricopeptide repeat (PPR) proteins in plant embryogenesis. Planta 221:424–436. doi:10.1007/s00425-004-1452-x
Deeba F et al (2012) Physiological and proteomic responses of cotton (Gossypium herbaceum L.) to drought stress. Plant Physiol Biochem 53:6–18. doi:10.1016/j.plaphy.2012.01.002
Deshmukh R et al (2014) Integrating omic approaches for abiotic stress tolerance in soybean. Front Plant Sci 5:244
Doerks T, Copley R, Bork P (2001) DDT- a novel domain in different transcription and chromosome remodeling factors. Trends Biochem Sci 26:145–146. doi:10.1016/S0968-0004(00)01769-2
Dubouzet JG et al (2003) OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J 33:751–763. doi:10.1046/j.1365-313X.2003.01661.x
Ford KL, Cassin A, Bacic A (2011) Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front Plant Sci 2:44. doi:10.3389/fpls.2011.00044
Gao S, Zhang YL, Yang L, Song JB, Yang ZM (2014) AtMYB20 is negatively involved in plant adaptive response to drought stress. Plant Soil 376:433–443. doi:10.1007/s11104-013-1992-6
Gelli M, Duo Y, Konda AR, Zhang C, Holding D, Dweikat I (2014) Identification of differentially expressed genes between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genom 15:179. doi:10.1186/1471-2164-15-179
Gharechahi J, Hajirezaei M-R, Salekdeh GH (2015) Comparative proteomic analysis of tobacco expressing cyanobacterial flavodoxin and its wild type under drought stress. J Plant Physiol 175:48–58. doi:10.1016/j.jplph.2014.11.001
Grandbastien M-A (2015) LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim Biophys Acta 1849:403–416. doi:10.1016/j.bbagrm.2014.07.017
Hadiarto T, Tran L-SP (2010) Progress studies of drought-responsive genes in rice. Plant Cell Rep 30:297–310. doi:10.1007/s00299-010-0956-z
Haefele SM, Kato Y, Singh S (2016) Climate ready rice: augmenting drought tolerance with best management practices. Field Crops Res 190:60–69. doi:10.1016/j.fcr.2016.02.001
Hald S, Nandha B, Gallois P, Johnson GN (2008) Feedback regulation of photosynthetic electron transport by NADP(H) redox poise. Biochim Biophys Acta 1777:433–440. doi:10.1016/j.bbabio.2008.02.007
Hancock RD et al (2014) Physiological, biochemical and molecular responses of the potato (Solanum tuberosum L.) plant to moderately elevated temperature. Plant Cell Environ 37:439–450. doi:10.1111/pce.12168
Hartwell LH, Hopfield JJ, Leibler S, Murray AW (1999) From molecular to modular cell biology. Nature 402:C47–C52
Hildebrandt T, Knuesting J, Berndt C, Morgan B, Scheibe R (2015) Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol Chem 396:523–537. doi:10.1515/hsz-2014-0295
Himmelbach A, Hoffmann T, Leube M, Höhener B, Grill E (2002) Homeodomain protein ATHB6 is a target of the protein phosphatase ABI1 and regulates hormone responses in Arabidopsis. EMBO J 21:3029–3038. doi:10.1093/emboj/cdf316
Hirochika H, Sugimoto K, Otsuki Y, Tsugawa H, Kanda M (1996) Retrotransposons of rice involved in mutations induced by tissue culture. PNAS 93:7783–7788
Huang X-S, Liu J-H, Chen X-J (2010) Overexpression of PtrABF gene, a bZIP transcription factor isolated from Poncirus trifoliata, enhances dehydration and drought tolerance in tobacco via scavenging ROS and modulating expression of stress-responsive genes. BMC Plant Biol 10:230. doi:10.1186/1471-2229-10-230
Huang X-S, Luo T, Fu X-Z, Fan Q-J, Liu J-H (2011) Cloning and molecular characterization of a mitogen-activated protein kinase gene from Poncirus trifoliata whose ectopic expression confers dehydration/drought tolerance in transgenic tobacco. J Exp Bot 62:5191–5206. doi:10.1093/jxb/err229
Izanloo A, Condon AG, Langridge P, Tester M, Schnurbusch T (2008) Different mechanisms of adaptation to cyclic water stress in two South Australian bread wheat cultivars. J Exp Bot 59:3327–3346
Jaresitthikunchai J, Phaonakrop N, Kittisenachai S, Roytrakul S (2009) Rapid in-gel digestion protocol for protein identification by peptide mass fingerprint. In: The 2nd biochemistry and molecular biology conference: biochemistry and molecular biology for regional sustainable development, Khon Kaen, May 7–8, 2009
Javid MG, Sorooshzadeh A, Sanavy SAMM, Allahdadi I, Moradi F (2011) Effects of the exogenous application of auxin and cytokinin on carbohydrate accumulation in grains of rice under salt stress. Plant Growth Regul 65:305–313
Ji K, Wang Y, Sun W, Lou Q, Mei H, Shen S, Chen H (2012) Drought-responsive mechanisms in rice genotypes with contrasting drought tolerance during reproductive stage. J Plant Physiol 169:336–344. doi:10.1016/j.jplph.2011.10.010
Jia Y et al (2015) Effect of low water temperature at reproductive stage on yield and glutamate metabolism of rice (Oryza sativa L.) in China. Field Crops Res 175:16–25. doi:10.1016/j.fcr.2015.01.004
Johansson C, Samskog J, Sundström L, Wadensten H, Björkesten L, Flensburg J (2006) Differential expression analysis of Escherichia coli proteins using a novel software for relative quantitation of LC–MS/MS data. Proteomics 6:4475–4485. doi:10.1002/pmic.200500921
Kant P, Kant S, Gordon M, Shaked R, Barak S (2007) STRESS RESPONSE SUPPRESSOR1 and STRESS RESPONSE SUPPRESSOR2, two DEAD-box RNA helicases that attenuate Arabidopsis responses to multiple abiotic stresses. Plant Physiol 145:814–830
Kapazoglou A, Drosou V, Argiriou A, Tsaftaris A (2013) The study of a barley epigenetic regulator, HvDME, in seed development and under drought. BMC Plant Biol 13:172
Kappachery S, Baniekal-Hiremath G, Yu JW, Park SW (2014) Effect of over-and under-expression of glyceraldehyde 3-phosphate dehydrogenase on tolerance of plants to water-deficit stress. Plant Cell Tissue Organ Cult 121:97–107. doi:10.1007/s11240-014-0684-0
Kawahara Y et al (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:1–10. doi:10.1186/1939-8433-6-4
Kosová K, Vítámvás P, Urban MO, Klíma M, Roy A, Prášil IT (2015) Biological networks underlying abiotic stress tolerance in temperate crops—a proteomic perspective. Int J Mol Sci 16:20913–20942. doi:10.3390/ijms160920913
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lisch D (2013) How important are transposons for plant evolution? Nat Rev Genet 14:49–61
Liu W et al (2014) The stripe rust resistance gene Yr10 encodes an evolutionary-conserved and unique CC-NBS-LRR sequence in wheat. Mol Plant 7:1740–1755. doi:10.1093/mp/ssu112
Lovisolo C, Perrone I, Carra A, Ferrandino A, Flexas J, Medrano H, Schubert A (2010) Drought-induced changes in development and function of grapevine (Vitis spp.) organs and in their hydraulic and non-hydraulic interactions at the whole-plant level: a physiological and molecular update. Funct Plant Biol 37:98–116
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Manna S (2015) An overview of pentatricopeptide repeat proteins and their applications. Biochimie 113:93–99. doi:10.1016/j.biochi.2015.04.004
Meierhoff K, Felder S, Nakamura T, Bechtold N, Schuster G (2003) HCF152, an Arabidopsis RNA binding pentatricopeptide repeat protein involved in the processing of chloroplast psbB-psbT-psbH-petB-petD RNAs. Plant Cell 15:1480–1495. doi:10.1105/tpc.010397
Meyer TS, Lamberts BL (1965) Use of coomassie brilliant blue R250 for the electrophoresis of microgram quantities of parotid saliva proteins on acrylamide-gel strips. Biochim Biophys Acta 107:144–145. doi:10.1016/0304-4165(65)90403-4
Morsy MR, Jouve L, Hausman J-F, Hoffmann L, Stewart JM (2007) Alteration of oxidative and carbohydrate metabolism under abiotic stress in two rice (Oryza sativa L.) genotypes contrasting in chilling tolerance. J Plant Physiol 164:157–167. doi:10.1016/j.jplph.2005.12.004
Nikolaeva MK, Maevskaya SN, Shugaev AG, Bukhov NG (2010) Effect of drought on chlorophyll content and antioxidant enzyme activities in leaves of three wheat cultivars varying in productivity. Russ J Plant Phys 57:87–95. doi:10.1134/s1021443710010127
Nounjan N, Siangliw JL, Toojinda T, Chadchawan S, Theerakulpisut P (2016) Salt-responsive mechanisms in chromosome segment substitution lines of rice (Oryza sativa L. cv. KDML105). Plant Physiol Biochem 103:96–105. doi:10.1016/j.plaphy.2016.02.038
Nouri M-Z, Moumeni A, Komatsu S (2015) Abiotic stresses: insight into gene regulation and protein expression in photosynthetic pathways of plants. Int J Mol Sci 16:20392–20416. doi:10.3390/ijms160920392
Oh M, Komatsu S (2015) Characterization of proteins in soybean roots under flooding and drought stresses. J Proteom 114:161–181. doi:10.1016/j.jprot.2014.11.008
Pandey V, Shukla A (2015) Acclimation and tolerance strategies of rice under drought stress. Rice Sci 22:147–161. doi:10.1016/j.rsci.2015.04.001
Park O-MK (2004) Proteomic studies in plants. BMB Rep 37:133–138
Perkins DN, Pappin DJC, Creasy DM, Cottrell JS (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567. doi:10.1002/(SICI)1522-2683(19991201)20:18<3551:AID-ELPS3551>3.0.CO;2-2
Pongprayoon W, Roytrakul S, Pichayangkura R, Chadchawan S (2013) The role of hydrogen peroxide in chitosan-induced resistance to osmotic stress in rice (Oryza sativa L.). Plant Growth Regul 70:159–173. doi:10.1007/s10725-013-9789-4
Pusnik M, Small I, Read LK, Fabbro T, Schneider A (2007) Pentatricopeptide repeat proteins in Trypanosoma brucei function in mitochondrial ribosomes. Mol Cell Biol 27:6876–6888. doi:10.1128/mcb.00708-07
Rai A, Singh R, Shirke PA, Tripathi RD, Trivedi PK, Chakrabarty D (2015) Expression of rice CYP450-like gene (Os08g01480) in Arabidopsis modulates regulatory network leading to heavy metal and other abiotic stress tolerance. PLoS One 10:e0138574. doi:10.1371/journal.pone.0138574
Saeed AI et al (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378
Saeng-ngam S, Takpirom W, Buaboocha T, Chadchawan S (2012) The role of the OsCam1-1 salt stress sensor in ABA accumulation and salt tolerance in rice. J Plant Biol 55:198–208. doi:10.1007/s12374-011-0154-8
Saikumar S, Varma CMK, Saiharini A, Kalmeshwer GP, Nagendra K, Lavanya K, Ayyappa D (2016) Grain yield responses to varied level of moisture stress at reproductive stage in an interspecific population derived from Swarna/O. glaberrima introgression line. NJAS Wagening J Life. doi:10.1016/j.njas.2016.05.005
Sakai H et al (2013) Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol 54:e6–e6. doi:10.1093/pcp/pcs183
Santoni V, Molloy M, Rabilloud T (2000) Membrane proteins and proteomics: un amour impossible? Electrophoresis 21:1054–1070. doi:10.1002/(SICI)1522-2683(20000401)21:6<1054:AID-ELPS1054>3.0.CO;2-8
Sato Y et al (2013) RiceFREND: a platform for retrieving coexpressed gene networks in rice. Nucleic Acids Res 41:D1214–D1221. doi:10.1093/nar/gks1122
Smita S, Katiyar A, Pandey DM, Chinnusamy V, BansaL KC (2015) Transcriptional regulatory network analysis of MYB transcription factor family genes in rice. Front Plant Sci. doi:10.3389/fpls.2015.01157
Sripinyowanich S et al (2013) Overexpression of a partial fragment of the salt-responsive gene OsNUC1 enhances salt adaptation in transgenic Arabidopsis thaliana and rice (Oryza sativa L.) during salt stress. Plant Sci 213:67–78. doi:10.1016/j.plantsci.2013.08.013
Tai F, Yuan Z, Wu X, Zhao P, Hu X, Wang W (2011) Identification of membrane proteins in maize leaves, altered in expression under drought stress through polyethylene glycol treatment. Plant Omics 4:250
Takahashi S, Murata N (2005) Interruption of the Calvin cycle inhibits the repair of Photosystem II from photodamage. Biochim Biophys Acta 1708:352–361. doi:10.1016/j.bbabio.2005.04.003
Takahashi S, Murata N (2006) Glycerate-3-phosphate, produced by CO2 fixation in the Calvin cycle, is critical for the synthesis of the D1 protein of photosystem II. Biochim Biophys Acta 1757:198–205. doi:10.1016/j.bbabio.2006.02.002
Tang X-M, Tao X, Wang Y, Ma D-W, Li D, Yang H, Ma X-R (2014) Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol Genet Genom 289:1075–1084. doi:10.1007/s00438-014-0869-6
Tefon BE, Maaß S, Özcengiz E, Becher D, Hecker M, Özcengiz G (2011) A comprehensive analysis of Bordetella pertussis surface proteome and identification of new immunogenic proteins. Vaccine 29:3583–3595. doi:10.1016/j.vaccine.2011.02.086
Thikart P, Kowanij D, Selanan T, Vajrabhaya M, Bangyeekhun T, Chadchawan S (2005) Genetic variation and stress tolerance of somaclonal variegated rice and its original cultivar. J Sci Res Chula Univ 30:63–75
Thorsell A, Portelius E, Blennow K, Westman-Brinkmalm A (2007) Evaluation of sample fractionation using micro-scale liquid-phase isoelectric focusing on mass spectrometric identification and quantitation of proteins in a SILAC experiment. Rapid Commun Mass Spectrom 21:771–778. doi:10.1002/rcm.2898
Tuteja N, Tarique M, Tuteja R (2014) Rice SUV3 is a bidirectional helicase that binds both DNA and RNA. BMC Plant Biol 14:283. doi:10.1186/s12870-014-0283-6
Tuteja N, Sahoo RK, Huda KMK, Tula S, Tuteja R (2015) OsBAT1 augments salinity stress tolerance by enhancing detoxification of ROS and expression of stress-responsive genes in transgenic rice. Plant Mol Biol Rep 33:1192–1209. doi:10.1007/s11105-014-0827-9
Udomchalothorn T, Maneeprasobsuk S, Bangyeekhun E, Boon-Long P, Chadchawan S (2009) The role of the bifunctional enzyme, fructose-6-phosphate-2-kinase/fructose-2,6-bisphosphatase, in carbon partitioning during salt stress and salt tolerance in Rice (Oryza sativa L.). Plant Sci 176:334–341. doi:10.1016/j.plantsci.2008.11.009
Udomchalothorn T, Plaimas K, Comai L, Buaboocha T, Chadchawan S (2014) Molecular karyotyping and exome analysis of salt-tolerant rice mutant from somaclonal variation. Plant Genome. doi:10.3835/plantgenome2014.04.0016
Umezawa T et al (2006) CYP707A3, a major ABA 8′-hydroxylase involved in dehydration and rehydration response in Arabidopsis thaliana. Plant J 46:171–182. doi:10.1111/j.1365-313X.2006.02683.x
Urano K, Kurihara Y, Seki M, Shinozaki K (2010) ‘Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr Opin Plant Biol 13:132–138. doi:10.1016/j.pbi.2009.12.006
Usadel B et al (2009) Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ 32:1633–1651. doi:10.1111/j.1365-3040.2009.02040.x
Vajrabhaya M, Vajrabhaya T (1991) Somaclonal variation for salt tolerance in rice. Biotechnol Agric For 14:368–382
Wang W-S et al (2011) Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot 62:1951–1960. doi:10.1093/jxb/erq391
Wang C, Yang Y, Wang H, Ran X, Li B, Zhang J, Zhang H (2016) Ectopic expression of a cytochrome P450 monooxygenase gene PtCYP714A3 from Populus trichocarpa reduces shoot growth and improves tolerance to salt stress in transgenic rice. Plant Biotechnol J 14:1838–1851. doi:10.1111/pbi.12544
Webster DE, Thomas MC (2012) Post-translational modification of plant-made foreign proteins; glycosylation and beyond. Biotechnol Adv 30:410–418. doi:10.1016/j.biotechadv.2011.07.015
Wessler SR (1996) Plant retrotransposons: turned on by stress. Curr Biol 6:959–961. doi:10.1016/S0960-9822(02)00638-3
Wolff S et al (2006) Gel-free and gel-based proteomics in Bacillus subtilis. Mol Cell Proteom 5:1183–1192. doi:10.1074/mcp.M600069-MCP200
Xiao X, Yang F, Zhang S, Korpelainen H, Li C (2009) Physiological and proteomic responses of two contrasting Populus cathayana populations to drought stress. Physiol Plant 136:150–168. doi:10.1111/j.1399-3054.2009.01222.x
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803. doi:10.1146/annurev.arplant.57.032905.105444
Yoo CY, Pence HE, Jin JB, Miura K, Gosney MJ, Hasegawa PM, Mickelbart MV (2010) The Arabidopsis GTL1 transcription factor regulates water use efficiency and drought tolerance by modulating stomatal density via transrepression of SDD1. Plant Cell 22:4128–4141. doi:10.1105/tpc.110.078691
Zavafer A, Cheah MH, Hillier W, Chow WS, Takahashi S (2015) Photodamage to the oxygen evolving complex of photosystem II by visible light. Sci Rep 5:16363. http://www.nature.com/articles/srep16363#supplementary-information
Zhang XM et al (2014) The cloning and characterization of a DEAD-box RNA helicase from stress-responsive wheat. Physiol Mol Plant Pathol 88:36–42. doi:10.1016/j.pmpp.2014.07.004
Zhu M, Chen G, Dong T, Wang L, Zhang J, Zhao Z, Hu Z (2015) SlDEAD31, a putative DEAD-box RNA helicase gene, regulates salt and drought tolerance and stress-related genes in tomato. PLoS One. doi:10.1371/journal.pone.0133849
Zörb C, Herbst R, Forreiter C, Schubert S (2009) Short-term effects of salt exposure on the maize chloroplast protein pattern. Proteomics 9:4209–4220. doi:10.1002/pmic.200800791
Acknowledgements
This work was supported by the Ratchadaphiseksompoj Research Fund, Chulalongkorn University (Grant No. CU-57-011-FW). NC was supported by the Science Achievement Scholarship of Thailand. MM was supported by DPST. OSBC was supported by CUAASC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Hajduch.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chintakovid, N., Maipoka, M., Phaonakrop, N. et al. Proteomic analysis of drought-responsive proteins in rice reveals photosynthesis-related adaptations to drought stress. Acta Physiol Plant 39, 240 (2017). https://doi.org/10.1007/s11738-017-2532-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-017-2532-4