
Abstract
Dihydroxyacetone synthase (DAS) and dihydroxyacetone kinase (DAK) are two key enzymes for formaldehyde assimilation in methylotrophic yeasts. In order to using a Gateway LR recombination reaction to construct a plant expression vector that contains the expression cassettes for the das and dak genes and allow the proteins encoded by the two target genes to be localized to the chloroplasts of transgenic plants, the entry vector pEN-L4*-PrbcS-*T-gfp-L3* contained the tomato rbcS 3C promoter (PrbcS) with its transit peptide sequence (*T) and a GFP reporter gene (gfp) was constructed in this study. To verify the applicability of pEN-L4*-PrbcS-*T-gfp-L3*, we generated an entry vector for the dak gene by replacing the gfp gene in this entry vector with the dak gene. We also generated an entry vector for the das gene by replacing the gus gene in another entry vector (pENTR*-PrbcS-*T-gus) with the das gene. Using these entry vectors and pK7m34GW2-8m21GW3, we successfully constructed the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus and the pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das expression vectors. Our results showed that high expression of GUS was achieved in leaves, and the expressed GFP, DAS and DAK proteins could be targeted to the chloroplasts after the two expression vectors were used to transform tobacco. The overexpressions of DAS and DAK in the chloroplasts successfully created a novel photosynthetic HCHO-assimilation pathway in transgenic tobacco. By utilizing these expression vectors, we not only successfully expressed two target genes with one transformation but also localized the expressed proteins to chloroplasts via the transit peptide sequence (*T). Therefore, the construction of pEN-L4*-PrbcS-*T-gfp-L3* establishes a technique platform that provides a convenient means for chloroplast genetic engineering.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Formaldehyde (HCHO) is thought to be a major air pollutant, a carcinogen and a cause of sick-house syndrome. Plants have been used to remediate HCHO pollution in an economical and environmentally conscious manner. However, the effect of this method is generally low due to the limited capacity of plants to metabolize HCHO (Schmitz et al. 2000). HCHO is produced during the early steps of methanol metabolism in microorganisms that can utilize methanol as a carbon source. Therefore, the cells of these organisms possess HCHO-assimilation pathways that can convert HCHO produced from C1 compounds into cellular constituents. A HCHO-assimilation pathway (ribulose monophosphate pathway) from a methylotrophic bacterium has been successfully incorporated into the Calvin cycle in transgenic plants in our recent studies (Chen et al. 2010; Song et al. 2010), which thereby enhances the ability of the transgenic plants to assimilate HCHO and remediate HCHO pollution. The xylulose monophosphate (XuMP) pathway is an HCHO-assimilation pathway in methylotrophic yeasts (Yurimoto et al. 2005). In this pathway, dihydroxyacetone synthase (DAS) catalyzes the first reaction in HCHO assimilation, which fixes HCHO to xylulose-5-phosphate (Xu5P) to produce dihydroxyacetone (DHA) and glyceraldehyde 3-phosphate (3PGA). DHA is toxic to yeast cells, and dihydroxyacetone kinase (DAK) is involved in the detoxification of DHA. The reaction catalyzed by DAK converts DHA to dihydroxyacetone phosphate (DHAP), which is non-toxic and can be utilized by yeast cells. Because Xu5P, DHAP and 3PGA are all intermediates of the Calvin cycle, we attempted to express yeast DAS and DAK in plant chloroplasts. By doing so, DAS can use the Xu5P generated in the Calvin cycle as the substrate to fix HCHO and produce DHA and 3PGA. The DHA can be phosphorylated by DAK to form DHAP. DHAP and 3PGA can re-enter the Calvin cycle as intermediates. Therefore, a novel HCHO-fixing pathway can be constructed with DAS and DAK (Fig. 1) in chloroplasts. This would enhance the ability of plants to uptake and metabolize HCHO and present a new strategy for the phytoremediation of HCHO pollution. However, the plant expression vectors for the das and dak genes cannot be constructed from the classical Agrobacterium binary vectors because they lack suitable restriction sites in the multiple cloning sites (MCSs).

Diagrams of the strategy for the installation of a novel photosynthetic HCHO-assimilation pathway by the overexpression of DAS and DAK in the chloroplasts of transgenic tobacco. Xu5P xylulose-5-phosphate, DHA dihydroxyacetone, DHAP dihydroxyacetone phosphate, [1- 13 C]3PGA [1-13C]glyceraldehyde 3-phosphate, [1- 13 C]DPGA [1-13C]1,3-diphosphoglyceric acid, [1- 13 C]PGAld [1-13C]3-phosphoglyceraldehyde, [3,4- 13 C]FBP [3,4-13C]fructose-1,6-bisphosphate, [3,4- 13 C]F6P [3,4-13C]fructose-6-phosphate, [3,4- 13 C]G6P [3,4-13C]glucose-6-phosphate, [3,4- 13 C]Gluc [3,4-13C]-glucose, [3,4- 13 C]Fruc [3,4-13C]fructose, RuBP ribulose 1,5-bisphosphate, E4P erythrose-4-phosphate, SBP sedoheptulose-1,7-bisphosphate, S7P sedoheptulose-7-phosphate
The Gateway cloning technology is developed based on the mechanism of site-specific recombination between the λ phage DNA and the Escherichia coli genome (Landy 1989). Gateway LR reaction is designed according to the excision strategy of the phage DNA from the E. coli chromosome. Construction of an expression vector via Gateway LR reaction for a target gene does not require the use of restriction enzymes and a ligase. The creation of the vector only requires the simple combination of an entry vector and a destination vector as well as the proteins required for integration. Therefore, Gateway cloning allows for the construction of an expression vector in an expeditious manner.
Recently, a collection of 36 Gateway entry clones has been developed to streamline the construction of the destination vectors used in gene functional analyses. The collection obeys the simple principle of genetic engineering. The genetic elements of the vectors are designed in a standard format and can be exchanged or combined depending on the requirements of researchers and the desired output. In addition, plant expression vectors containing MultiSite Gateway recombination sites have been constructed. Two or three genes of interest can be subcloned simultaneously into the separate expression cassettes of these expression vectors (Karimi et al. 2007). For example, the attR1-ccdB-attR2 expression cassette is located between the ROLD promoter and OCS terminator, and the attR4-ccdB-attR3 cassette is located between the CaMV35S (35S) promoter and terminator in the destination vector pK7m34G2-8m21GW. Thus, the DNA fragments of the entry vectors containing attL1-gene1-attL2 and attL4-gene2-attL3 can be integrated simultaneously in this binary vector via one Gateway LR reaction to produce a plant expression vector that contains two target gene expression cassettes.
The Gateway destination vector pK7m34G2-8m21GW3 is suitable for the construction of a plant expression vector that contains the expression cassettes carrying the das and dak genes. However, few restriction sites can be used for gene insertion downstream of the attL4 recombination site of all currently available commercial entry vectors containing attL4-gene2-attL3 fragments. Moreover, there are no chloroplast transit peptide sequences behind the 35S and ROLD promoters in the pK7m34GW2-8m21GW3 destination vector. Therefore, the current commercial entry vectors and the destination vector pK7m34GW2-8m21GW3 cannot be used directly to construct a plant expression vector that carries the expression cassettes for these two target genes and localizes the expressed target proteins to chloroplasts.
To address the problem described above, a modified entry vector pEN-L4*-PrbcS-*T-gfp-L3* containing the light inducible promoter (PrbcS) and a transit peptide sequence (*T) as well as a GFP reporter gene (gfp) was constructed in the present study. In order to verify the applicability of pEN-L4*-PrbcS-*T-gfp-L3*, the entry vector pEN-L4*-PrbcS-*T-dak-L3* was constructed from this entry vector for the dak gene. Similarly, the pENTR*-PrbcS-*T-das for the das gene was constructed from the entry vector pENTR*-PrbcS-*T-gus (Ma et al. 2011) containing attL1 and attL2 recombination sites. Using these entry vectors and the destination vector pK7m34GW2-8m21GW3 to perform Gateway LR reactions, the plant expression vector pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus containing the expression cassettes of two reporter genes (gfp and gus) and the plant expression vector pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das containing the expression cassettes of das and dak genes were constructed. These two plant expression vectors were used to transform tobacco to examine whether these genes can be expressed normally to allow the expressed proteins to be targeted into chloroplasts and functioned.
Materials and methods
Bacterial strains and plasmids
E. coli DH5α (Invitrogen) was used for DNA construction and plasmid amplification.
Candida boidinii S2 (1950) was purchased from CICC (China Center of industrial Culture Collection). Pichia pastoris GS115 (his4) was purchased from Invitrogen. The Gateway entry vector pEN-L4-2-L3 and the plant destination vector pK7m34GW2-8m21GW3 were purchased from VIB/Gent (Belgium). Agrobacterium strain C58C1 (pMP90) was used for plant transformation.
Site-directed mutagenesis
Mutagenesis was conducted in pEN-L4-2-L3 using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s instructions. The mutagenic sense primer HindIII5 (5′-GGTGACACTATAGAAG*C*T*TCAAGCTATGCA-3′) and the mutagenic antisense primer HindIII3 (5′-TGCATAGCTTGAA*G*C*TTCTATAGTGTCACC-3′) were used to introduce the HindIII site into pEN-L4-2-L3; the resulting vector was named pEN-L4*-2-L3. Similarly, the mutagenic sense primer XhoI5 (5′-TACTATTCTAGTCGACCTC*G*AGGCGGCCGCACTAGTGA-3′) and the mutagenic antisense primer XhoI3 (5′-CTACTAGTGCGGCCGCCTC*G*AGGTCGACTAGAATAGTA-3′) were used to convert the PstI site to an XhoI site in pEN-L4*-2-L3 to produce the entry vector pEN-L4*-2-L3*. Asterisks indicate the mutation sites. Following mutation, the correct plasmid sequences were verified by restriction enzyme digestion and sequence analysis.
TA cloning
The coding regions of das (AF086822, 2121 bp) and dak (AF019198, 1827 bp) genes were amplified by PCR using the genomic DNA prepared from C. boidinii S2 and P. pastoris GS115 (his4) as templates, respectively. For construction of the Gateway entry vectors, the coding region of das gene was amplified with sense primer 5′-gcatgcCTCTCGCAAAAGCTGCTTC-3′ (containing a SphI site) and antisense primer 5′-ggatccTTATTGATCATGTTTTGGTTTTTC-3′ (containing a BamHI site, the sequence (AATCATTTA) encoding the signal peptide for peroxisome destination was removed and not included in the antisense primer to avoid the targeting of the expressed DAS into peroxisomes in transgenic plants); the coding region of dak gene was amplified with sense primer 5′gcatgcTCTAGTAAACATTGGG-3′ (containing a SphI site) and antisense primer 5′- ctcgagCTACAACTTGGTTTCAGATTTG-3′ (containing an XhoI site). The amplified PCR products were subcloned into a pMD18-T vector (TaKaTa, Japan) to generate TA cloning vectors, pMD18-TA1-das and pMD18-TA1-dak, respectively. For construction of prokaryotic expression vectors, the coding region of das gene was amplified with sense primer 5′-catatgGCTCTCGCAAAAGCTGCTTC-3′ (containing an NdeI site) and antisense primer 5′-ggatccTTGATCATGTTTTGGTTTTTC-3′ (containing a BamHI site); the coding region of dak gene was amplified with sense primer 5′-catatgTCTAGTAAACATTGGGATTAC-3′ (containing an NdeI site) and antisense primer 5′-ctcgagCAACTTGGTTTCAGATTTGAAG-3′ (containing an XhoI site). The amplified PCR products were subcloned into a pMD18-T vector (TaKaTa, Japan) to generate TA cloning vectors, pMD18-TA2-das and pMD18-TA2-dak, respectively. Following TA cloning, the correct plasmid sequences were verified by restriction enzyme digestion and sequence analysis.
Gateway LR reaction
Entry vectors and the destination vector pK7m34GW2-8m21GW3 were purified using a plasmid purification kit. The Gateway LR Clonase plus Enzyme Mix (Invitrogen) was used to perform LR reactions according to the manufacturer’s instructions. The reaction mixture was incubated overnight at 25 °C and transformed into E. coli DH5α competent cells. The recombination clones were selected on LB-medium plates with 50 μg/ml spectinomycin (Spe).
Plant transformations
The plant expression vectors were transferred into A. tumefaciens C58C1 (pMP90) by electroporation using Gene Pulser (BioRad) with parameters of 200 ohms and 2.5 kV/0.2 cm. A. tumefaciens transformants were selected on LB-agar plates containing 100 μg/ml Spe. Tobacco (Nicotiana tabacum cv. Xanthi) was transformed following the leaf disk co-cultivation protocol of Horsch et al. (1985).
Growth and maintenance of transgenic plants
For aseptic growth of tobacco, the resistant shoots were first selected on Murashige and Skoog (MS) medium with Kanamycin (Kan, 50 μg/ml) and then transferred to MS medium (pH 5.7) containing sucrose (3 %) and grown in a growth chamber at 25 °C under constant light. For the growth on soil, the plantlets with roots were first selected on Kan-containing medium and then transplanted to pots with 1/2 perlite plus 1/2 organic soil.
Observation of GFP fluorescence and GUS staining analysis
Observation of GFP fluorescence was performed as described by Ma et al. (2011) using a confocal laser-scanning microscope (FV1000S, Olympus, Japan) with Kr/Ar laser excitation. Fresh leaves of tobacco were transversely sectioned and immediately mounted between a glass slide and a cover slip. GFP and chlorophyll fluorescence were induced at an excitation wavelength of 488 nm, and detected at emission wavelengths of 500–515 nm and 630–680 nm, respectively. GUS staining analysis was performed as described by Zhao et al. (2001) with mature leaves from tobacco plants grown aseptically on MS medium.
Expression of DAS and DAK recombinant proteins and preparation of antibodies
The DNA fragment containing the das coding region was obtained by digesting pMD18-TA2-das with the NdeI and BamHI enzymes. The fragment was then ligated into the pET-28a(+) vector between the NdeI and BamHI sites to generate the pET-28a-das prokaryotic expression vector. The DNA fragment containing the dak coding region was obtained from pMD18-TA2-dak digested with the NdeI and XhoI enzymes and ligated into pET-28a(+) between the NdeI and XhoI sites to generate the pET-28a-dak prokaryotic expression vector.
For expressions of the DAS and DAK recombinant proteins, pET-28a-das and pET-28a-dak were introduced into E. coli BL21. The DAS recombinant protein was expressed in inclusion bodies of E. coli BL21 and recovered from the polyacrylamide gel after the inclusion body protein was dissolved and separated by SDS-PAGE. The DAK recombinant protein was expressed as soluble protein in E. coli BL21 and purified with a HisTrap HP (5 ml) column (Amersham Biosciences, Sweden). The purification was performed by following the instructions of the manufacturer. Mice were immunized with the purified DAS and DAK recombinant proteins, and the DAS and DAK antibodies were prepared from the immunized mouse sera and used for Western blot analysis.
Western blot analysis
Leaves (5 g) of tobacco plants grown on agar medium were harvested. Intact chloroplasts were isolated from tobacco leaves as described by Miyagawa et al. (2001). The chloroplasts were disrupted and centrifuged to obtain the soluble protein fraction. For Western blot analysis, proteins were separated in SDS-PAGE gel (12 %) and then transferred to polyvinylidene difluoride (PVDF) membranes by electroblotting. The membranes were first treated with mouse antibodies raised against DAS or DAK and then with a goat antibody raised against mouse IgG which had been conjugated with peroxidase.
H13CHO and NaH13CO3 labeling experiments
For H13CHO labeling experiments, aseptic tobacco leaves (3 g) were treated in 70 ml of a 2 mM H13CHO (Cambridge Isotopes Laboratories, Cambridge, MA, USA) solution (containing 0.1 % MES (w/v), pH 5.7). For NaH13CO3 labeling experiments, aseptic tobacco leaves (3 g) were soaked in 70 ml of a 5 mM NaH13CO3 (Cambridge Isotopes Laboratories, Cambridge, MA, USA) solution (containing 0.1 % MES (w/v), pH 5.7). The plants were incubated under constant light (100 μmol m−2 s−1) at 25 °C for 30 min with shaking (100 rpm). After incubation, the plants were washed with cooled sterile water 5 times to remove free H13CHO or NaH13CO3 from the leaf surfaces and then quickly frozen in liquid nitrogen. The frozen leaves were ground into a fine powder with a mortar and pestle and extracted with 3 ml of a 100 mM potassium phosphate buffer (KPB, pH 7.4) containing 10 mM maleic acid (used as an internal reference). After thawing, the extract was boiled for 3 min and centrifuged at 12,000 rpm for 10 min to remove cellular debris. The supernatant was frozen, lyophilized and resuspended in 0.5 ml of 100 mM KPB. The extract was then transferred into a 5-mm NMR tube and subjected to 13C-NMR analysis.
13C-nuclear magnetic resonance analysis
13C-NMR analysis was performed on a Bruker DRX 500-MHz instrument (Bruker Biosciences Corporation, Billerica, MA). 13C-NMR data were collected using parameters as described by Chen et al. (2010). The acquisition parameters included a 5-μs (90°) pulse with broadband proton decoupling, a spectral width of 37,594 Hz, an acquisition time of 0.5 s, and a decay time of 1.2 s. The sample temperature was maintained at 25 °C, and 32,000 data points were acquired for each sample. Twelve hundred scans were acquired for each sample, and a line broadening of 4 Hz was used in the processing of the data. Chemical shifts were obtained by reference to maleic acid at 130.41 ppm. The resonances were assigned based on the chemical shifts of authentic samples. To calculate the relative contents of the metabolites, the target peaks were integrated relative to maleic acid at 130.41 ppm.
Gaseous HCHO-uptake analysis
The measurement for the gaseous HCHO uptake rate by tobacco plants was performed in a 294-l glass chamber (700 × 600 × 700 mm) as described by Song et al. (2010). The light (75 μmol m−2 s−1) was supplied by lamps on two sides of the chamber. Air-polluting HCHO was introduced by a pump or injected into the chamber using a syringe. The gaseous HCHO was circulated through four small fans in the corner of the chamber. The plant pot, covered with plastic bags, was placed into the chamber. The gaseous HCHO concentration in the chamber was detected using a gaseous HCHO sensor (CH2O/C-10, Membrapor, Switzerland), which was connected to a monitor. The measurements were initiated when the HCHO concentration was close to 4 ppm. To maintain HCHO in a gaseous state, the temperature and the humidity of the chamber were controlled at 26–32 °C and 25–60 %, respectively, over the entire measurement period. After the measurements had been taken, all of the leaves were collected from the plants. The leaf areas were measured as described by Song et al. (2010).
Results
Construction of the entry vectors
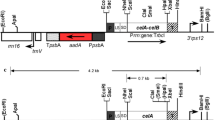
The Gateway entry vector pEN-L4-2-L3 was used as the backbone for construction of the entry vector pEN-L4*-PrbcS-*T-gfp-L3*. To subclone the PrbcS-*T-gfp DNA fragment (2.4 kb) of the Gateway entry vector pENTR*-PrbcS-*T-gfp, which we constructed previously (Ma et al. 2011), into the entry vector pEN-L4-2-L3 between the attL4 and attL3 sites, an HindIII site was introduced downstream of the attL4 site in pEN-L4-2-L3 via site-directed mutagenesis to produce pEN-L4*-2-L3. Next, the PstI site in pEN-L4*-2-L3 was converted into an XhoI site by the same method to produce the modified entry vector pEN-L4*-2-L3*. The PrbcS-*T-gfp fragment was subsequently excised from pENTR*-PrbcS-*T-gfp and introduced into pEN-L4*-2-L3* between the HindIII and XhoI sites to produce the entry vector pEN-L4*- PrbcS-*T-gfp-L3* (Fig. 2a). In this entry vector, three restriction enzyme sites (HindIII, NdeI and SalI) were present upstream of the PrbcS promoter; there is an NcoI and a SphI site in the 5′ end and 3′ end of the *T transit peptide sequence, respectively; the MCS at the 3′ end of the gfp gene contained eight restriction sites (BamHI, SmaI, XmaI, KpnI, SacI, EcoRI, NotI and XhoI). To test the applicability of the pEN-L4*-PrbcS-*T-gfp-L3* entry vector, the dak gene obtained from pMD18-TA1-dak was subcloned between the SphI and XhoI sites to replace the gfp gene in this entry vector, producing pEN-L4*-PrbcS-*T-dak-L3* (Fig. 2b). Similarly, the das gene obtained from the pMD18-TA1-das vector was subcloned between the SphI and BamHI sites to replace the gus gene in the pENTR*-PrbcS-*T-gus entry vector, producing pENTR*-PrbcS-*T-das (Fig. 2c).

Schematic diagrams of the pEN-L4*-PrbcS-*T-gfp-L3* (a), pEN-L4*-PrbcS-*T-dak-L3* (b) and pEN-L4*-PrbcS-*T-das-L3* (c) constructs. attL1, attL2, attL3 and attL4, four specific attachment sites for the Gateway LR reaction; kan r the kanamycin resistance gene, PrbcS the tomato rbcS-3C promoter, *T the tomato rbcs-3C transit peptide sequence with the NcoI site overlapping with the ATG initiation codon, gfp the protein-coding region of green fluorescent protein, das the protein-coding region of dihydroxyacetone synthase, dak the protein-coding region of dihydroxyacetone kinase
Construction of plant expression vectors and the generation of transgenic plants
To generate the plant expression vector pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus (Fig. 3a), pEN-L4*-PrbcS-*T-gfp-L3*, pENTR*-PrbcS-*T-gus and the pK7m34GW2-8m21GW3 destination vector were used to perform an LR reaction. In this reaction, the PrbcS-*T-gfp segment from pEN-L4*-PrbcS-*T-gfp-L3* and the PrbcS-*T-gus segment from pENTR*-PrbcS-*T-gus were subcloned between the attB4/attB3 and the attB2/attB1 sites, respectively, in the generated plant expression vector. Similarly, pEN-L4*-PrbcS-*T-dak-L3*, pENTR*-PrbcS-*T-das and the pK7m34GW2-8m21GW3 destination vector were used to perform another LR reaction to yield the plant expression vector pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das (Fig. 3b). In this reaction, the PrbcS-*T-dak segment from pEN-L4*-PrbcS-*T-dak-L3* and the PrbcS-*T-das segment from pENTR*-PrbcS-*T-das were subcloned between the attB4/attB3 and attB2/attB1 sites, respectively, in the yielded plant expression vector. In the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus and pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das expression vectors, the expressions of gfp, gus , das and dak, along with their respective transit peptide sequences, were directly regulated by the tomato rbcS-3C promoter PrbcS. To verify the functions of these two expression vectors, we transformed tobacco with pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus and pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das to generate transgenic tobacco plants. We obtained 15 transgenic lines (named SP1-SP15) for pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus and 20 transgenic lines (named SK1-SK20) for pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das, respectively.

Diagrams of the T-DNA constructs in the plant expression vectors. The constructs a and b were inserted into the Gateway destination vector pK7m34GW2-8m21GW3. gus, the protein-coding region of β-glucuronidase; gfp, das and dak are described in Fig. 2; LB and RB the left and right border sequences, respectively, NPT II the kanamycin resistance gene, P35S CaMV 35S promoter sequences, attB1, attB2, attB3 and attB4 four specific attachment sites for the Gateway BP reaction, t35S the CaMV 35S terminator sequence, PROLD an Agrobacterium rhizogenes promoter sequence, NosT the nopaline synthase terminator sequence
Expression of GUS in the leaves and localization of GFP in the chloroplasts of transgenic tobacco
Histochemical GUS-staining analysis were conducted to determine if GUS was expressed in the leaves of pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus transgenic tobacco. The third leaf (from the top) of several tobacco plants grown on MS agar medium was selected for GUS-staining analysis. Figure 4a and b show representative results for the stained leaves. The leaf of the transgenic line (Fig. 4b) was extensively stained, whereas the WT leaf remained unstained (Fig. 4a). The data indicated that the GUS protein was expressed in the leaves of the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus transgenic tobacco.

GUS expression and chloroplast localization of GFP in the transgenic tobacco leaves. a The WT leaf, b the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus leaf, c–e the WT leaf, f−h the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus leaf, c and f red chlorophyll fluorescence, d and g green GFP fluorescence, e and h two fluorescence images were overlaid. Bars 5 μm
To confirm the subcellular localization of the expressed GFP protein, we examined the distribution patterns of GFP fluorescence in leaf mesophyll cells of the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus transgenic lines under a confocal laser-scanning microscope. In WT plants, chlorophyll autofluorescence (red) was observed at emission wavelengths greater than 560 nm, but no signal was detected for GFP at emission wavelengths of 505–530 nm (Fig. 4c–e). In pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus mesophyll cells, GFP fluorescence (green) and chlorophyll autofluorescence were co-localized in chloroplasts (Fig. 4f–h). These observations suggested that the expressed GFP protein encoded by GFP gene from the pKm-35S-PrbcS-*T-gfp-PROLD-PrbcS-*T-gus vector could be targeted directly into the chloroplast of transgenic tobacco leaves.
Expression of DAS and DAK in the chloroplasts of transgenic tobacco
The genomic insertion of the das/dak genes and the transcription of the two genes were investigated in 10 of the 20 kan-resistant transgenic tobacco lines by genomic PCR and RT-PCR analysis, respectively. The results indicated that the das and dak genes from the pKm-35S-PrbcS-*T-dak-PROLD-PrbcS-*T-das vector were integrated normally into the genomes of the transgenic tobacco lines and transcribed (data not shown). Moreover, all of the transgenic plants exhibited a normal phenotype when grown on soil or MS medium. Three transgenic lines (SK3, SK5 and SK6) were selected for subsequent experiments.
The expression of the das and dak genes in the chloroplasts of the three selected lines was examined by Western blot analysis with mouse antibodies against the DAS (Fig. 5a) and DAK (Fig. 5b) recombinant proteins. Chloroplasts were purified by subcellular fractionation. The DAS and DAK proteins were detected in the soluble protein fraction of the chloroplast extracts of the three transgenic lines but not in the extracts of the chloroplasts of the negative control. The removal of the transit peptide in the chloroplasts was verified by the molecular sizes of DAS (77.4 kDa) and DAK (66.7 kDa), which were identical to those of the endogenous proteins expressed in C. boidinii S2 and P. pastoris G115 (his4) yeast cells, respectively. These results indicate that the DAS and DAK proteins were successfully expressed and targeted to the chloroplasts of the transgenic tobacco.

Western analysis for detection of the DAS (a) and DAK (b) protein expression in the chloroplasts of the transgenic tobacco plants. Western analysis was performed as described in the “Materials and methods” section. The DAS antibody (a) and the DAK antibody (b) were used to detect the expression of the DAS and DAK proteins, respectively; 20 μg of soluble protein from chloroplasts was loaded in each lane. Crude protein (5 μg), prepared from cells of C. boidinii S2 and P. pastoris GS115 (his4), were used as positive controls. WT the extract from the chloroplasts of wild-type tobacco leaves, SK5, SK3 and SK6 the extracts from the chloroplasts of three transgenic tobacco lines, DAK and DAS positive controls
H13CHO metabolism in transgenic SK tobacco
When DAS was expressed in E. coli, it was present in inclusion body as no active form. A 13C-NMR technique was used to analyze the H13CHO metabolic spectrum in the transgenic SK5 line to confirm that the overexpressed DAS and DAK proteins were active in vivo and functioned to allow the installation of an HCHO-assimilation pathway in chloroplasts. As the installed HCHO-assimilation pathway couples with Calvin-Benson cycle (Fig. 1), it can be predicted that the metabolic profile of H13CHO-assimilation via the installed DAS/DAK pathway in the transgenic tobacco is similar to that of NaH13CO3-assimilation through the Calvin cycle in WT plants since the same metabolic intermediates are produced in the two pathways. DAS catalyzes the first reaction of the DAS/DAK pathway to allow H13CHO to be directly fixed to Xu5P, producing DHA and [1-13C]3PGA. DAK catalyzes the phosphorylation of DHA to generate DHAP, which can enter the Calvin cycle. Because DHAP is not labeled by 13C, its production will not be monitored by the 13C-NMR analysis. The [1-13C]3PGA produced from the H13CHO assimilation is expected to have the same metabolic fate as that produced in the first step (catalyzed by Rubisco) of NaH13CO3-assimilation via the Calvin cycle. During this reaction, [1-13C]3PGA is first reduced to [1-13C]1, 3-diphosphoglyceric acid ([1-13C]DPGA). Next, a part of [1-13C]DPGA is isomerized to generate [1-13C]DHAP, [3, 4-13C]fructose -1, 6-bisphosphate ([3, 4-13C]FBP) is then synthesized by the condensation of [1-13C]DPGA and [1-13C]DHAP. The dephosphorylation of [3,4-13C]FBP forms [3,4-13C]fructose-6-phosphate ([3,4-13C]F6P), which can be isomerized to generate [3,4-13C]glucose-6-phosphate ([3,4-13C]G6P). [3, 4-13C]G6P can be converted to free [3, 4-13C]fructose ([3, 4-13C]Fruc) and [3, 4-13C]glucose ([3, 4-13C]Gluc) or used for the synthesis of cell constituents. Due to the low abundance and fast turnover in plants, it is not easy to observe the resonance peaks of most metabolic intermediates, whereas free fructose and glucose are present in plants at high levels to allow easy detection for their resonance peaks by the 13C-NMR analysis. Because the third and fourth carbon atoms ([3,4-13C]) of these sugars are labeled first in the tracer experiment, the enhancements in the signal peaks corresponding to the two carbon atoms will be observed before those of the other fructose and glucose carbon atoms.
In the 13C-tracer experiments, the leaves of the transgenic SK5 plants were treated with H13CHO. The leaves of the WT tobacco were treated with H13CHO and NaH13CO3. An extract of WT tobacco leaves without treatment was used as the control (CK) to monitor the background 13C-NMR signal levels of the intermediates, which are shown in Fig. 6d. The 13C-NMR spectrum (Fig. 6b) of the SK5 leaves labeled with H13CHO was compared with those of the WT leaves labeled with NaH13CO3 (Fig. 6a) and H13CHO (Fig. 6c). The metabolic profile of H13CHO in the transgenic SK5 line was almost identical to that of NaH13CO3 in the WT tobacco, except for the presence of a resonance peak (with a chemical shift at 170.94 ppm), corresponding to formate (H13COOH), which was produced by the oxidation of H13CHO. The 13C-NMR spectrum of WT leaves labeled with H13CHO showed no significant difference from that of the CK control, except the resonance signal peak for H13COOH. This verified that the metabolic pathway for H13CHO assimilation in the transgenic SK5 tobacco is similar to that for NaH13CO3 assimilation in the WT tobacco. H13CHO was assimilated via DAS/DAK pathway in addition to being oxidized to H13COOH in transgenic tobacco. However, in WT leaves, H13CHO was only oxidized to H13COOH, and no assimilation was observed. As we predicted, the strengths of resonance signal peaks corresponding to [3-13C]Fruc, [4-13C]Fruc, [3-13C]Gluc and [4-13C]Gluc with chemical shifts at 79.87, 75.06, 75.16 and 69.98 ppm, respectively, in the SK5 spectrum were similar to those of WT leaves treated with NaH13CO3, and all enhanced as compared with those in the WT spectrum treated with H13CHO. The relative integrals (Fig. 6e, f) of these signal peaks were 1.7, 1.5, 1.6 and 1.5 times of those in the H13CHO-treated WT leaves, respectively. This indicates that the expressed DAS/DAK were active in vivo and functioned in transgenic plants to allow effectively fixation of H13CHO through the Calvin cycle to generate more [3, 4-13C]Fruc and [3, 4-13C]Gluc.

13C-NMR analysis of H13CHO and NaH13CO3 metabolite profiles in transgenic and WT tobacco leaves. Complete 13C-NMR spectra are shown on left side, expanded regions of interest are shown on right side. a The extract of WT leaves treated with NaH13CO3. b The extract of SK5 leaves treated with H13CHO. c The extract of WT treated with H13CHO. d The extract of WT leaves without treatment. 13C-labeling and 13C-NMR analysis are performed as described in “Materials and methods” section. e Relative integrals of [3-13C]Fruc and [4-13C]Fruc signal peaks. f Relative integrals of [3-13C]Gluc and [4-13C]Gluc signal peaks. Peak assignments are as follows, Ref maleic acid, H 13 COOH formate, [3- 13 C]Fruc [3-13C] fructose, [4- 13 C]Fruc [4-13C] fructose, [3- 13 C]Gluc [3-13C] glucose, [4- 13 C]Gluc [4-13C] glucose
Augmentation of the purification capacity for HCHO-polluted air in transgenic SK tobacco
To confirm that the overexpressed DAS/DAK pathway will enhance the capacity of transgenic SK tobacco plants to purify HCHO-polluted air, we measured the uptake rate of gaseous HCHO by transgenic tobacco plants (Fig. 7). As shown in Fig. 7, the time required for WT, SK3, SK5 and SK6 plants to consume 50 % of the initial HCHO was 40, 20, 15 and 15 min, respectively. To remove all of the gaseous HCHO, the time required for SK3, SK5 and SK6 plants were 50, 50 and 45 min, respectively. HCHO still remained at 0.8 ppm in the chamber after the WT plants were placed in the chamber for 55 min. The uptake rates of gaseous HCHO for WT, SK3, SK5 and SK6 plant leaves were 0.48, 0.68, 0.80 and 0.71 ppm m−2 min−1, respectively. These results indicate that the purification capability of the transgenic plants was higher than that of WT plants. Thus, the installation of the DAS/DAK pathway into tobacco plants increased their ability to remove gaseous HCHO from the atmosphere.

Kinetics of gaseous HCHO elimination by transgenic tobacco plants. The tobacco plants were grown in pots in a greenhouse for 1–2 months, and plants of equivalent size were used for the analysis. The experiment was repeated more than three times, and representative results are shown. CK, the soil-containing pot without plants
Discussion
With the development of plant genetic engineering, manipulation of a single gene has become more and more easy. However, many important characteristics and metabolic pathways of plants are involved in functions of multiple genes. Therefore, genetic engineering must be performed to manipulate multiple genes at a time. Traditional methods for the introduction of multiple genes into plant genomes include sexual crossing, continuous transformation and the simultaneous transformation of many plasmids. However, these methods are time-consuming and laborious. Lin et al. (2003) developed a vector system that could assemble multiple gene expression cassettes into a plant expression vector. The system consists of a receptor vector which was constructed based on a transformation-competent artificial chromosome (TAC) and two donor vectors. However, the donor vectors do not contain promoter and terminator sequences. Thus, the users cannot directly subclone the coding regions of their target genes into the donor plasmids, which is inconvenient for the application of this system. Goderis et al. (2002) constructed six auxiliary (pAUX) vectors with six homing endonucleases and a binary vector with an MCS containing the six homing endonucleases. The expression cassettes of the six target genes can be assembled into this binary vector using the six homing endonucleases. Chung et al. (2005) transferred the pAUX vectors into a modular system to develop pSAT vectors. pSAT vectors contain different promoter and terminator sequences and an extended MCS. With these vectors, one can substitute for the promoter and terminator sequences or subclone target genes into the pSAT vectors between the promoter and terminator sequences. Using the six homing endonucleases, six expression cassettes of target genes can be assembled into the Agrobacterium binary vector pPZP-RCS1 or pPZP-RCS2 (Tzfira et al. 2005). However, assembly of the six expression cassettes from the pSAT vectors into the binary vectors still requires restriction digestion and ligation. Therefore, subcloning in this system is laborious and time-consuming.
The MultiSite Gateway cloning system allows the assembly of multiple DNA fragments in a predefined order, direction and framework without restriction digestion and ligation. Recently, a number of binary vectors containing multiple recombination sites have been developed. Using these vectors, the users can easily interchange the promoter, coding region and terminator sequence of a gene (Karimi et al. 2007). However, the use of a transit peptide to target the protein of interest into chloroplasts requires the initiation codon sequence and the transit peptide sequence to be in the same reading frame. The exchange of the DNA sequence in the binary destination vector depends on its flanking recombination sites. However, even the shortest recombination site is at least 20 bp long. Therefore, the MultiSite Gateway cloning technology cannot be used directly for constructing plant expression vectors that target proteins of interest into chloroplasts.
Recently, the MultiSite Gateway technology has been developed for the simultaneous cloning of multiple gene expression cassettes in a versatile format. In this study, an HindIII site was introduced downstream of the attL4 site of the pEN-L4-2-L3 entry vector, and the PstI site upstream of the attL3 site of this plasmid was converted to an XhoI site by site-directed mutagenesis, generating the pEN-L4*-2-L3* vector. The PrbcS-*T-gfp fragment from pENTR*-PrbcS-*T-gfp vector (Ma et al. 2011) was subcloned into the pEN-L4*-2-L3* vector by restriction digestion/ligation to construct the pEN-L4*-PrbcS-*T-gfp-L3* entry vector with a size of only 5.0 kb. A SphI site is present in the initiation codon region of gfp, and the MCS in the 3’ end of gfp contained eight restriction sites (BamHI, SmaI, XmaI, KpnI, SacI, EcoRI, NotI and XhoI). If the SphI site and one of the eight restriction sites described above is added to the 5′ end and 3′ end of a target gene (x), respectively, the DNA fragment of x gene can be digested, excised and subcloned into pEN-L4*-PrbcS-*T-gfp-L3* to replace gfp and produce the entry vector pEN-L4*-PrbcS-*T-x-L3 for the gene x. Similarly, another target gene (y) can be used to replace the gus gene in the entry vector pENTR*-PrbcS-*T-gus (5.8 kb), which contains an attL1 site and an attL2 site, to produce the entry vector pENTR*-PrbcS-*T-y for the gene y. Because the sizes of the two entry vectors are small, it is not difficult to subclone the target gene into these entry vectors using a restriction digestion/ligation strategy. Consequently, the plant expression vector containing the expression cassettes of x and y genes can be constructed with the two entry vectors pEN-L4*-PrbcS-*T-x-L3* and pENTR*-PrbcS-*T-y as well as the destination vector pK7m34GW2-8m21GW3 via a Gateway LR reaction. The genetic manipulation of the two target genes (x and y) can be achieved through a transformation event. The expressions of the x and y genes are regulated by the PrbcS promoter. The expressed target proteins encoded by the x and y genes can also be targeted to the chloroplasts of the transgenic plants via the transit peptide sequence (*T). These two entry vectors allow researchers to conveniently construct light-inducible expression vectors for any target gene for the genetic engineering of metabolic pathways in chloroplasts. If the *T transit peptide sequence is removed by NcoI digestion from the entry vector, no transit peptide sequence exists in the final plant expression vector. Thus, the expression of the target gene is still under the control of the PrbcS promoter, but the expressed target protein will remain in the cytoplasm. In this manner, the application of Gateway technology and the destination vectors can be further expanded.
Chen et al. (2010) successfully established a photosynthetic HCHO-assimilation pathway in transgenic Arabidopsis and tobacco plants via the overexpression of 3-hexulose-6-phosphate synthase (HPS) and 6-phospho-3-hexuloisomerase (PHI), two key enzymes in a HCHO assimilation pathway in a methylotrophic bacterium, in chloroplasts, which thereby enhanced the capacity of the transgenic plants for HCHO absorption and assimilation. Song et al. (2010) overexpressed the HPS/PHI fusion protein in the chloroplasts of transgenic geranium (Pelargonium sp. Frensham) to create the same photosynthetic HCHO-assimilation pathway. As a result, the transgenic geranium remediated the HCHO-polluted air more effectively than the WT geranium. In this study, a plant expression vector containing expression cassettes for the das and dak target genes from methylotrophic yeasts was successfully constructed with the pENTR*-PrbcS-*T-gus and pEN-L4*-PrbcS-*T-gfp-L3 entry vectors. Consequently, transgenic tobacco could be generated via introduction of the two target genes through a transformation event. Our data showed that both the das and dak genes were inserted normally into the genomes of the transgenic plants and transcribed in the transgenic tobacco leaves. The DAS and DAK proteins were also expressed and successfully targeted to the chloroplasts of the transgenic plants. The H13CHO labeling experiment confirmed that the expressed DAS and DAK proteins were active and functional. The uptake rate of gaseous HCHO from the polluted air by the transgenic SK tobacco was approximately 40 % faster than that of WT tobacco. This is an improvement over previous studies, which showed that the uptake rate of gaseous HCHO by the transgenic tobacco plants overexpressing HPS and PHI and the transgenic geranium overexpressing the HPS/PHI fusion protein were ~20 % (Chen et al. 2010) and 25 % (Song et al. 2010) faster than that of the WT plants, respectively. Therefore, the overexpression of DAS and DAK in the chloroplasts constructed an alternative photosynthetic HCHO-assimilation pathway in transgenic plants, which provides a new strategy for genetic engineering to improve the phytoremediation capacity for HCHO-pollution in transgenic plants.
Author contribution
K.-Z. Li and L.-M. Chen designed research; S.-Q. Xiao, Z. Sun, S.-S. Wang and J. Zhang performed research; S.-Q. Xiao and L.-M. Chen analyzed data and wrote the paper.
References
Chen LM, Yurimoto H, Li KZ, Orita I, Akita M, Kato N, Sakai Y, Izui K (2010) Assimilation of formaldehyde in transgenic plants due to the introduction of the bacterial ribulose monophosphate pathway genes. Biosci Biotechnol Bioch 74:627–635
Chung SM, Frankman EL, Tzfira T (2005) A versatile vector system for multiple gene expression in plants. Trends Plant Sci 10:357–361
Goderis IJ, De Bolle MF, François IE, Wouters PF, Broekaert WF, Cammue BP (2002) A set of modular plant transformation vectors allowing flexible insertion of up to six expression units. Plant Mol Biol 50:17–27
Horsch RB, Fry JE, Hoffmann NL, Eichholtz D, Rogers SG, Fraley RT (1985) A simple and general method for transferring genes into plants. Science 227:1229–1231
Karimi M, Depicker A, Hilson P (2007) Recombinational cloning with plant gateway vectors. Plant Physiol 145:1144–1154
Landy A (1989) Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu Rev Biochem 58:913–949
Lin L, Liu YG, Xu XP, Li BJ (2003) Efficient linking and transfer of multiple genes by a multigene assembly and transformation vector system. Proc Natl Acad Sci USA 100:5962–5967
Ma L, Song ZB, Hu QQ, Zhao Y, Nian HJ, Yu YX, Li KZ, Chen LM (2011) Construction and application of a Gateway entry vector with rubisco small subunit promoter and its transit peptide sequence*. Prog Biochem Biophys 38:269–279
Miyagawa Y, Tamoi M, Shigeoka S (2001) Overexpression of a cyanobacterial fructose-1, 6-sedoheptulose-1, 7-bisphosphatase in tobacco enhances photosynthesis and growth. Nat Biotechnol 19:965–969
Schmitz H, Hilgers U, Weidner M (2000) Assimilation and metabolism of formaldehyde by leaves appear unlikely to be of value for indoor air purification. New Phytol 147:307–315
Song ZB, Orita I, Yin F, Yurimoto H, Kato N, Sakai Y, Izui K, Li KZ, Chen LM (2010) Overexpression of an HPS/PHI fusion enzyme from Mycobacterium gastri in chloroplasts of geranium enhances its ability to assimilate and phytoremediate formaldehyde. Biotechnol Lett 32:1541–1548
Tzfira T, Tian GW, Lacroix B, Vyas S, Li J, Leitner-Dagan Y, Krichevsky A, Taylor T, Vainstein A, Citovsky V (2005) pSAT vectors: a modular series of plasmids for autofluorescent protein tagging and expression of multiple genes in plants. Plant Mol Biol 57:503–516
Yurimoto H, Kato N, Sakai Y (2005) Assimilation, dissimilation, and detoxification of formaldehyde, a central metabolic intermediate of methylotrophic metabolism. Chem Rec 5:367–375
Zhao Y, Christensen SK, Fankhauser C, Cashman JR, Cohen JD, Weigel D, Chory J (2001) A role for flavin monooxygenase-like enzymes in auxin biosynthesis. Science 291:306–309
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (Grant # 30670163 to L.M.C.) and by the Foundation for Yunnan Province and Kunming University of Science and Technology for Training Adult and Young Leaders of Science and Technology in Yunnan (Grant # 2004PY01-5 to L.M.C.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Moriguchi.
Rights and permissions
About this article
Cite this article
Xiao, SQ., Sun, Z., Wang, SS. et al. Overexpressions of dihydroxyacetone synthase and dihydroxyacetone kinase in chloroplasts install a novel photosynthetic HCHO-assimilation pathway in transgenic tobacco using modified Gateway entry vectors. Acta Physiol Plant 34, 1975–1985 (2012). https://doi.org/10.1007/s11738-012-0998-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11738-012-0998-7