
Abstract
The drug loading efficiency of magnetite nanoparticles (MNPs) can be enhanced by coating with polyethylene glycol (PEG) which is a biocompatible polymer. The PEG-coated MNPs could be the potential candidates for carrying the drug molecules to the targeted sites. In this study, size-controlled MNPs were synthesized and functionalized with PEG of molecular weights 700, 2000 and 5000. The MNPs and PEGylated MNPs (PMNPs) samples were characterized through X-ray diffraction (XRD), thermogravimetric analysis (TGA), Fourier transform infrared (FTIR) spectroscopy, transmission electron microscopy (TEM) and surface area and pore size measurements by Brunauer, Emmett and Teller (BET) and Barrett–Joyner–Halenda (BJH) methods. The diffraction data showed that MNPs are purely crystalline with face cubic crystal structure, whereas the spherical shape of the particles was confirmed by TEM. The TGA supports thermal stability of nanoparticles which was markedly enhanced by coating with PEG. The BJH data (hysteresis loops) showed that MNPs were mesoporous in nature. After characterization, the PEGylated MNPs were loaded with gallic acid (GA). The spectroscopic evidences regarding the successful PEGylation and loading of GA onto PMNPswere acquired from FTIR spectroscopy. The in vitro sustained drug release efficacy of PMNPs was evaluated via UV–visible spectroscopy. Among all the synthesized samples, P750MNPs-10 showed the highest % drug release i.e., 98% into phosphate buffer saline (PBS) solution of pH 4.4 and 90% in PBS of pH 7.4. The highest % drug release at pH 10 may be attributed to smaller particle size with high surface area. The highest % drug release can also be associated with the weak interactions between P750MNPs-10 and GA through hydrogen bonding.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanotechnology is an emerging research technology in modern science which is widely used in the pharmaceutical and electronic industries as well as for controlled drug release to et al. 2017). The use of nanomaterials offers many advantages due to their specific physicochemical properties which differ from the bulk material (Faraji et al. 2010). A useful type of magnetic material is the magnetite nanoparticles (MNPs) which have been widely applied for targeted drug delivery from last many years. It was reported that coating of MNPs has enhanced their stability and are considered as most effective for multiple applications due to their paramagnetic behavior (Prijic and Sersa 2011). Further, MNPs were effectively used for controlled drug release to treat cancer cell line (Cho et al. 2008; De Jong and Borm 2008). The nanotechnology has been used to tackle the challenges such as unspecific uptake by other organs, low capability of tissue penetration and decrease in effective concentration due to dilution in the body fluids (Allen and Cullis 2013; Allen et al. 1995). These challenges are, however, overcome by using nanocarriers which are compatible to drug in the cellular drug delivery system, where the cells act as drug carrier due to membrane fluidity and interact with the other tissue due to biocompatibility (Tan et al. 2015).
For the biomedical applications, iron oxide nanoparticles are encapsulated in a polymeric coating, which provide these particles stability and compatibility (Bulte and Kraitchman 2004). Iron during metabolism, can be recycled from the iron oxide particles (Arruebo et al. 2007; Chomoucka et al. 2010). The superparamagnetic characters of MNPs are essential for the drug delivery because it prevents the agglomeration in the capillary vessel (Plank et al. 2003). The polymer coating of MNPs makes them superior, increases ability to target specific cells, and decrease the toxic effect toward healthy cells and tissue. The coating of MNPs with neutrals or hydrophilic compounds such as PEG, PVA and polysaccharide has increased the circulatory half-life from minutes to hours (Mahmoudi et al. 2008,2009; Paul et al. 2004). Further, the coated MNPs have novel cancer therapeutic vector due to cheap and easy preparation, sustained drug release properties, better encapsulation properties, biocompatible with normal cells and high magnetic properties. It was also reported by various researchers that the agglomeration of MNPs can be prevented by coating with polymers (Barahuie et al. 2017; Zbigniew et al. 2017; Dorniani et al. 2013). The polymers with simple chemical structure contain more hydroxyl groups which are suitable for the biomedical applications due to nontoxicity, biocompatibility and non-carcinogenicity, etc.
Different methods were developed for the in vitro drug release study. In the sample and separate method, loaded particles were introduced into release media (buffer saline solution) at constant temperature, while the concentration of drugs was measured at regular intervals by taking out appropriate amount of sample from release media and by adding equal amount of fresh buffer saline solution to maintain the sink condition. The sample and separate technique is reasonably straight forward and simple, but sometime aggregation of NPs occurs during drug release study if experiment was not handled carefully (D’Souza 2014). The continuous flow method is another technique to monitor drug release study by using USP IV apparatus. However, this approach has many disadvantages like cost of instrument, difficulties in setup and filter clogging (D’Souza 2014). Dialysis method has also been used to assess the drug release from the nanoparticles by using dialysis bag, but this method is not simple and straightforward (D’Souza 2014). The modern synthetic method develops architecture for the synthesis of conjugate system of polymer and NPs for drug loading in sophisticated and controllable manner (covalent and non-covalent approaches). The non-covalent approach is simple and easy method for loading/releasing of drug, while covalent method enables drug release only by external stimuli (Ulbrich et al. 2016).
The nanoparticles were employed as nanocarriers for many drugs in previous work but failed due to their toxicity, less biocompatibility and non-degradability inside the body. Therefore, to resolve this issue, researchers have developed innovative conjugative systems where nanohydroxy particles as a core were coated with PEG and wrapped with insulin or gallic acid. In the conjugated system (using non-covalent approach), it was found that PEG has increased the hydrophilicity, which in turn protects nanoparticles from degradation in the body. The PEG-functionalized nanocomposites are overall safe, nontoxic for drug delivery and are effective for sustained drug release due to hydrogen bonding between PEG and gallic acid (Zhang et al. 2018). The size-controlled MNPs have never been studied, where effect of different molecular weights of PEG on the drug release efficacy was investigated. The objectives of the present work were, therefore, to study the effect of size-controlled MNPs and their coating with PEG by various molecular weights on the loading and release properties of GA.
In the present work, sample and separate method was used to assess the in vitro drug release, while drug-loaded onto NPs was done via non-covalent interaction. The pH-controlled MNPs-10, MNPs-11, MNPs-12 were fabricated by microemulsion-assisted co-precipitation method which is useful to control and tailor the size of MNPs. The P750MNPs-11, P750MNPs-12, P750MNPs-12, P2000MNPs-10, P2000MNPs-11, P2000MNPs-12, P5000MNPs-10, P5000MNPs-11, P5000MNPs-12 were synthesized and loaded by gallic acid (GA). The synthesized MNPs were coated with different molecular weight of PEG (750, 2000 and 5000) to investigate the effect of hydroxyl group on the loading and release efficacy of GA. After PEGylation and GA loading, the particles were characterized with state-of-the-art tools. The experiments have shown that the size of MNPs and chain length of coated polymer affect the drug release efficacy. The study, therefore, showed the importance of size and selectivity of polymer that can play a critical role in sustained drug release. In this study, it was concluded that size of nanoparticles and molecular weight of PEG have affected the release behavior of GA. Such affects may be useful in future study to perform in vivo study of GA and in vitro and in vivo release behavior of other drugs for biomedical applications.
Materials and methods
Chemicals
All the chemicals and materials were of analytical grade purchased from Sigma-Aldrich. Polyethylene glycols (99.6%) of molecular weights 750, 2000 and 5000 were used for PEGylating the MNPs. Deionized water was used throughout the experiments. The gallic acid was purchased from Sigma-Aldrich with 99.5% purity.
Synthesis of magnetite nanoparticles
The MNPs were prepared by microemulsion-assisted co-precipitation method which is a novel technique as has already been employed in our previously published paper (Khan et al. 2019). 50 mL of an aqueous solution of each iron (III) chloride hexahydrate (5 mM) and iron (II) chloride tetrahydrate (2.5 mM) were added to a preformed microemulsion prepared by mixing 0.07 mL of n-heptane and 100 mL of 5 mM (0.014 g/100 mL) sodium dodecyl sulfate (SDS, 99.9%) with vigorous stirring. Each reaction was carried out separately at pH 10, 11 and 12. The reaction mixture was stirred at 60 °C till the formation of black precipitate which indicates the formation of MNPs. After aging for 48 h, the precipitates were separated with the aid of a permanent magnet and washed repeatedly with ethanol and deionized water. Finally, it was calcined at 250 °C for 24 h and stored in polymeric vials for further study.
Coating of MNPs with PEG
First of all, 0.6 g of MNPs was added in 0.01 g PEG of molecular weight 700 (already dissolved in 100 mL DI water) and stirred for 5 h. Then 0.1 M ammonium hydroxide solution (10 mL) was added to aid condensation of the PEG onto MNPs. After 48 h, the coated MNPs were washed with DI water to remove unreacted PEG. Finally, the PEG-coated MNPs were dried in oven at 60 °C. Similar procedure was adopted for coating MNPs with the PEG of molecular weights 2000 and 5000.
Drug encapsulation and release study of GA by PMNPs
Loading was achieved by suspending PMNPs in 0.2 g/mL of GA solution (prepared in 80:20 methanol and DI water) for 10 h followed by washing and oven-drying at 60 °C. The drug encapsulation of PEGylated magnetite nanoparticles was investigated by using
The drug release efficiency of PMNPs was noted at 37 °C by adding 0.02 g of nanoparticles to 10 mL of phosphate buffer saline (PBS) solutions at pH 7.4. After appropriate time interval, 3.0-mL aliquot was removed with the addition of fresh PBS to achieve the required sink conditions. The concentration of GA was measured by using Optizen 3220 UV spectrophotometer using a quartz cell (path length 0.10 cm) at a wavelength of 275 nm (extinction coefficient, ε = 4.20 dm3 mol−1 cm−1). Similar drug release experiments were performed by taking PBS solution at pH 4.4.
Characterization of MNPs and coated MNPs
The FTIR spectra of MNPs were recorded on a Perkin Elmer Spectrum 100 FTIR spectrometer operating between 500 and 4000 cm−1 at a resolution of 0.04 cm−1. The phase composition of MNPs and PMNPs was determined by using XRD technique. Elemental analyses and the morphology of the nanoparticles were investigated by SEM/EDX with an Oxford Instrument INCA X-Act 10 mm2 silicon drift detector. TEM was performed by using the model JEOL 2100 microscope with a LaB6 at 200 kV bottom mounted Ruby camera. Thermal stability was investigated in thermogravimetric analyzer model Setsys Evolution TGA 16/18* from Setaram. The program *Calisto* is employed to collect and process the data. The sample is loaded into a 170-µL alumina crucible. The temperature program consists of a heating range 30–700 °C at 10 K/min. The experiments are performed under argon flow at 20 mL/min. The BET surface areas were measured by nitrogen adsorption at 77 K using a 3Flex unit from Micromeritics.
Results and discussion
Surface area and pore size distribution
The adsorption–desorption isotherms of the PMNPs are shown in Fig. 1 and displayed type IV behavior. H3 type of hysteresis loop further confirms their mesoporous structure (Guerra et al. 2009). This hysteresis loop is usually found on precipitated MNPs and indicates slit-shaped pores (Khan et al. 2019). It was observed that the surface area decreased when the MNPs were synthesized at higher pHs. The MNPs prepared at lower pH showed particles of smaller diameters and less agglomerated. The position of the P/Po inflection fell in the range of 0.8–1 which point toward the mesoporous structure of PMNPs. The pore size volume distribution was calculated using BJH method (Khan et al. 2019). The pore diameter of P5000MNPs-10 was observed at around 30 nm to 40 nm calculated by distribution peak (Fig. 1a), although there were a small number of larger pores. A similar, though slightly larger at around 32- to 40-nm distribution, was found for the P5000MNPs-11 (Fig. 1b). The pore volume of P5000MNPs-12 was again slightly larger (Fig. 1c). It has also been inferred that at high pressure (P/Po > 0.99) capillary agglomeration occurred, and the isotherm increased rapidly to form a loop. The surface areas were measured using the BET method, and the extracted data is shown in Table 1.

N2 adsorption/desorption isotherms and pore size distribution of PMNPs synthesized at a pH 10; b pH 11; c pH 12
FTIR spectroscopy
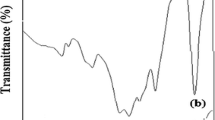
The FTIR spectra of these particles (MNPs-10, MNPs-11, MNPs-12) are shown in Fig. 2a. The absorption band around 3000–3500 cm−1 and weak absorption band at 1107 cm−1 is assigned to the stretching vibration of OH groups. A weak vibration band at 570 cm−1 and 903 cm−1 can be associated to be the bending vibration of Fe and oxygen particles (Pang et al. 2011). The absorption bands at the 960 cm−1 and 2955 cm−1 may be present due to CH and CH2 out-of-plane bending vibrations in a PEG (Fig. 2b). The absorption bands present at 1240 cm−1 and 1465 cm−1 are associated due to the stretching vibration of OH group (Sun et al. 2006b)_ENREF_22. The absorption bands present at 3250 and 1016 are assigned to stretching vibrations of OH group and C = O in gallic acid (Fig. 2c). The main absorption bands in the drug-loaded PMNPs samples are due to GA, e.g., the sharp bands appeared at 1570 cm−1 and 1370 cm−1 associated with the COO stretching. The FTIR spectra of MNPs after loading with PEG and GA, showed the success of the loading method (Fig. 2d–f). The absorption bands around 1090 cm−1 and 1110 cm−1 are attributed to the presence of Fe–O–C (Kayal and Ramanujan 2010). The bands appeared at 990 cm−1 and 2960 cm−1 can be assigned to CH2 and CH out-of-plane bending vibrations due to PEG on the surface of MNPs (Kayal and Ramanujan 2010). The weak absorption bands at 1240 cm−1 and 1400 cm−1 are due to stretching vibration of OH group in the gallic acid (Sun et al. 2006a). The spectra from MNPs prepared at different pHs were similar, indicating similar functional groups, but showed differences in the % transmission. The change in % transmission is correlated with the particle size and hence to the relative amounts of PEG.

FTIR spectra of pure MNPs (a), PEG (b), GA (c) and GPMNPs synthesized at different pH (d–f)
X-ray diffraction analysis (XRD)
The diffraction peaks of GA-loaded PMNPs (Fig. 3) were found matched with the reference cards of MNPs possessing face-centered cubic crystalline structures. The diffraction peaks at 2θ were recorded 30.25°, 35.61°, 43.42°, 53.59°, 57.32° 62.70o for the GP750MNPs-10, while the 30.21°, 35.57°, 43.17°, 53.63°, 57.15° 62.26° were observed for the GP750MNPs-11 and 30.21°, 35.57°, 43.17°, 53.56°, 57.08°, 62.73°, 74.11° for GP750MNPs-12. These diffractions were matched with the reference card 01-0741-4918. All the diffractions were related to characteristics peaks of MNPs; however, a little shift in the absorption intensities was noted by varying the pH which may be due to different ionic strength. The diffraction peaks (30.28°, 35.61°, 43.31°, 53.63°, 57.23°, 62.81°, 74.41°) at 2θ were observed for the GP2000MNPs-10. These diffractions were found matched well when compared to reference cards (01-0741-4918) and (01-071-6336). The diffraction peaks of GP2000MNPs-11 and GP2000MNPs-12 were appeared at 30.25°, 35.61°, 43.20°, 53.59°, 57.23°, 62.12°, 62.81°, 74.19° and 30.21°, 35.57°, 43.17°, 53.48°, 57.12°, 62.71°, 74.19°), respectively (Ref. card: 01-0741-4918), whereas for the GP5000MNPs-10, these diffraction peaks (27.05°, 30.25°, 35.57°, 43.17°, 53.85°, 57.08°, 62.73°, 74.11°) were matched to the same reference card as mentioned above. It was observed that no significant diffraction peaks were appeared due to GA or PEG. The XRD also allowed estimation of the particle sizes of the nanoparticles by using Scherrer formula.
where λ = is the wavelength of X-ray (0.15 cm) B = full width at half maximum (FWHM), θ = corresponding Bragg angle and K (0.89) is shape parameter. The variations in size of GA-loaded PMNPs were observed as the particles synthesized at three different pHs have different sizes. The sizes for GP750MNPs-10, GP750MNPs-11, GP750MNPs-12 were calculated 34.2 nm, 34.6 nm and 34.9 nm, respectively. The size estimated for the GP2000MNPs-10, GP2000MNPs-11, GP200 GP5000MNPs-100MNPs-12 were 34.4 nm, 34.8 nm and 35.2 nm, respectively. However, the crystallite sizes for the samples GP5000MNPs-10, GP5000MNPs-11, GP5000MNPs-12 were found 44.8 nm, 45.1 nm and 45.7 nm, respectively.

XRD of GPMNPs (Cerda-Sumbarda et al. 2019) coated with different molecular weight of PEG at different pH
TEM images of MNPs and GA-loaded PMNPs
The size and shape of the nanoparticles were investigated using TEM. Figure 4 shows micrographs of the bare MNPs-10, MNPs-11, and MNPs-12. The particles were found spherical, and the sizes estimated (using ImageJ-32 software) for MNPs-10 MNPs-11, MNPs-12 were around 2–8 nm, 10–15 nm, 12–35 nm, respectively. A significant amount of agglomeration was observed in the sample prepared at highest pH. The process of coating with PEG and then gallic acid, has increased the particle size in the samples GP750MNPs-10, GP2000MNPs-10 and GP5000MNPs-10 and was measured around 10–15 nm while in the samples GP750MNPs-11, GP2000MNPs-11 and GP5000MNPs-11 it was around 20–25 nm. Similarly, the particle size in GP750MNPs-12, GP2000MNPs-12 and GP5000MNPs-12 was in the range 23–50 nm. From the particle size data, it can be concluded that higher pH was found responsible for the increased size. Further, incorporation of gallic acid also enhanced the agglomeration of particles, particularly with the higher molecular weight of PEG (Fig. 5).

TEM images of GA-loaded PMNPs synthesized at different pH (10–12) with different molecular weights of PEG

The average size of MNPs calculated by using ImageJ-32 software
In the present study, microemulsion-assisted co-precipitation method was used to synthesize MNPs where difference in the size can be explained based on sizes of nanodroplets and concentration of reactants (Rehman et al. 2019). Here, the controlled size of particles was achieved through the pH and ionic strength of the aqueous phase (Curl 1963; Niemann et al. 2006,2008; Niemann and Sundmacher 2010). This increase in size can be explained based on growth model concept which explained the free ions in solution that are thermodynamically stable clusters (LaMer and Dinegar 1950). The growth of NPs was caused by change in solubility of precursor in solution. The high solubility and surface energy of smaller particles within solution, re-dissolve these particles and result in the formation of larger particles that will grow further (Thanh et al. 2014). The pH of the solution plays an effective role in controlling the size of particles which has increased the negative charge at the interface. Such negative charge has provided double-negative layer and facilitated the process of agglomeration and hence resulted an increase in size of particles at higher pH. The size of nanoparticles varies with the ionic strength of precipitating medium. The surface parameters show, at higher pH, more strength of precipitating agent in the sense of ionic strength, which cause increase in the size of nanoparticles (Darminto et al. 2011).
The increase in size of PMNPs is due to the presence of more OH groups present on the surface of PEG and the higher molecular weight of PEG causes more agglomeration and resulting increased in size. From the TEM images it is concluded that the non-uniform in size and shape of GA-loaded PMNPs were investigated, which may be due to the agglomeration of smaller particles. The size variation of MNPs was determined by using ImageJ-32 software which shows that while encapsulation by PEG makes the particles are more stable. Moreover, it has been demonstrated that the PEG provided higher stability to MNPs as it prevents agglomeration of individual particles due to surface coating (Saifuddin et al. 2009).
TG/DTG analysis
The thermal stabilities of GA-loaded PMNPs were characterized by recording weight loss curves as shown in Fig. 6. The first weight loss at 120 °C may be assigned to removal of physically adsorbed water (Vuković et al. 2009), whereas the gradual weight loss at 227 °C to 253 °C may be assigned due to weight loss GA (Dorniani et al. 2012). Another weight loss at 350 °C may also be assigned to the decomposition of PEG. The weight loss at such a higher temperature could be due to interaction of hydrogen bonding with already retained OH groups on the surface of MNPs. The thermal changes noted above 350 °C may be due to decomposition of some amount of PEG from the surface of MNPs along with the phase changes at such higher temperatures (Dorniani 2014). Further, different weight losses were detected for MNPs by altering the pH and molecular weights of PEG. This investigation shows that the GPMNPs are thermally stable. The stability of gallic acid-loaded PMNPs enhanced due to hydrogen bonding between gallic acid and PEG on the surface of MNPs. The different degradation behaviors as evaluated for GPMNP for particles synthesized at different pH (10–12) were due to different molecular weight of PEG.

TG/DTG of GA-loaded P (750, 2000, 5000) MNPs (A-I) fabricated at different pH10-12
In case of GP750MNPs-10, three stages of weight losses were detected. The first weight loss was recorded at 120 °C, while the second (5%) and third weight losses (0.02%) at 120–260 °C and 330 °C, respectively, could be assigned to the breakdown of intermolecular forces of different physiosorbed molecules on the surface or in interstitial space and decomposition of GA and PEG. The thermogram of GP750MNPs-11 showed the gradual weight loss of 1.5%, 2.5% p and 0.03%. Similarly, the weight losses detected in the sample GP750MNPs-12 were 1% (120 °C), 5.2% and 0.04% at 120–320 °C and at 350 °C, respectively. The weight loss gradually increases for the samples GP2000M-10, GP2000MNPs-11, GP2000MNPs-12 and GP5000MNPs-10, GP5000MNPs-11, GP5000MNPs-12 with increased molecular weight of PEG. The very small weight loss above 350 °C may be due to decarboxylation of –COOH functional group of GA attached to the external wall of PEG through hydrogen bonding (Tang et al. 2006). The weight loss of about 2.5% (682 °C) may be due to phase transition of MNPs. The comparative analysis from the result of TGA/DTG shows that GA-loaded P750MNPs-10 was the most thermally stable among all the studied samples.
Drug encapsulation/release efficiency of MNPs
The encapsulation efficiency of MNPs synthesized at different pH with different molecular weights of PEG was evaluated. The drug encapsulation efficiency was found in between 5 and 8%. In P750MNPs-10 it was 8% while for samples P2000MNPs-10, P5000MNPs-10, P750MNPs-11, P2000MNPs-11, P5000MNPs-11, P750MNPs-12, P2000MNPs-12 and P5000MNPs-12 it was 7.8%, 7.2%, 7.4%, 7.69%, 7.1%, 7.3%, 5.5% and 6.7%, respectively. The in vitro % release of GA was investigated into two different phosphate buffer solutions at pH 7.4 and 4.4 at 37 °C as shown in Fig. 7 (Cerda-Sumbarda et al. 2019). The amount of drug release varied with time and reached a maximum about 61% within almost 800 min at pH 7.4 and 90% at pH 4.4. The pH of the buffer was chosen according to the pH of blood and the cell where pKa value of GA is 4 (Table 2).

% GA released from the P750 MNPs (a), P2000 MNPs (b) and P5000 MNPs (c) in PBS (pH = 4.4 and 7.4) solutions
The observed drug release efficiency was found higher in the buffer having pH 4.4 than 7.4, which may be due to degradation of self-aggregated NPs in strong acid release media (4.4). This inferred that gallic acid molecules were hydrolyzed with the interaction of acid due to hydrogen bonding (Tao et al. 2016). The sustained release of GA from the surface of PMNPs may be due to the presence of strong hydrogen bonding between gallic acid and polyethylene glycol, so the PMNPs are the most suitable candidates for release of drugs molecules inside the cell. The pH has played an important role in drug release study. In present study, the drug release in pH 4.4 was found higher, and this finding as has also been reported in previous studies, is vital for cancer treatment (Na et al. 2003, 2007). Secondly, it is close to lysosomal acid lipase pH which is around 4.4 and its best activity was shown at 4.4 pH which decreases to 60% when the pH was changes from 4.4.
The highest release of GA (98% and 90%) at both pH values was observed by the sample P750MNPs-10. The release of GA was gradually decreased for the particles synthesized at higher pH. The higher release may be due to the higher surface area of P750MNPs-10 which is expected to bind more GA than the other PMNPs. The highest GA release can also be associated with the presence of fewer hydroxyl groups present on the surface of low molecular weight PEG as compared with PEG of higher molecular weights where strong intermolecular forces could be the reason of slow release of GA. Therefore, based on this observation, MNPs-10 synthesized and functionalized with lower molecular weight of 750 may be useful for the in vitro sustained release of GA. The release kinetics of GA from the surface of nanoparticles can be analyzed using various models. In the present study, pseudo-second-order model (Eq. 2) was found well fitted to the data (Fig. 8) (Dong et al. 2010).
where qt and qe are the equilibrium release rate at time “t” and equilibrium release amount. The R2 values obtained from the straight line trend further inferred the applicability of pseudo-second-order reaction for the release of GA (Tang, Dou, and Sun). Table 3 represents the detailed kinetic parameters for the release of GA from PMNPs. The % GA release was also compared with the literature, and it was found higher than the reported values (Tang, Dou and Sun). The drug release comparison is shown in Table 4.

Pseudo-second-order reaction for the release of GA from PMNPs into PBS solutions (7.4 and 4.4)
Conclusions
The microemulsion-assisted co-precipitation method was used to synthesized MNPs as well as PMNPs. MNPs with a range of sizes have been successfully synthesized by varying the solution pH and then coated with PEG of varying chain length. The TEM confirms that the particles were spherical and are totally pH controlled. The diffraction data show that MNPs were well crystalline with face-centered cubic (FCC) geometry. The thermal study (TGA/DTA) confirms that gallic acid-loaded PEGylated magnetite nanoparticles are stable and suitable for in vitro studies. The mesoporosity was evaluated for the synthesized PMNPs using BJH method, while the highest BET surface area was noted for the particles fabricated at pH 10. It was confirmed that PMNPs were successfully loaded with GA whose release kinetics followed pseudo-second-order model. Among all the synthesized sample, GP750MNPs-10, showed the highest (98%) and sustained release of GA into PBS at pH 4.4. It has been inferred that both size and shape of nanoparticles were found affective in the release efficiency of gallic acid.
References
Allen TM, Cullis PR (2013) Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev 65(1):36–48. https://doi.org/10.1016/j.addr.2012.09.037
Allen TM, Hansen CB, de Menezes DEL (1995) Pharmacokinetics of long-circulating liposomes. Adv Drug Deliv Rev 16(2):267–284. https://doi.org/10.1016/0169-409X(95)00029-7
Arruebo M, Pacheco R, Ibarra M, Santamaría J (2007) Magnetic nanoparticles for drug delivery. Nano Today 2:22–32. https://doi.org/10.1016/S1748-0132(07)70084-1
Barahuie F, Dorniani D, Saifullah B, Gothai S, Hussein MZ, Pandurangan AK, Arulselvan P, Norhaizan ME (2017) Sustained release of anticancer agent phytic acid from its chitosan-coated magnetic nanoparticles for drug-delivery system. Int J Nanomed 12:2361–2372. https://doi.org/10.2147/IJN.S126245
Bulte JWM, Kraitchman DL (2004) Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed 17(7):484–499. https://doi.org/10.1002/nbm.924
Cerda-Sumbarda YD, Zapata-Gonzalez I, Licea-Claverie A, Zizumbo-Lopez A, Ramos-de Valle LF, Espinoza-Martínez A (2019) Poly(hexylacrylate)Core-poly(ethyleneglycol methacrylate)Shell nanogels as fillers for poly(2-hydroxyethyl methacrylate) nanocomposite hydrogels. Polym Eng Sci 59(1):170–181. https://doi.org/10.1002/pen.24884
Cho K, Wang X, Nie S, Chen Z, Shin DM (2008) Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res 14(5):1310–1316. https://doi.org/10.1158/1078-0432.ccr-07-1441
Chomoucka J, Drbohlavova J, Huska D, Adam V, Kizek R, Hubalek J (2010) Magnetic nanoparticles and targeted drug delivering. Pharmacol Res 62(2):144–149. https://doi.org/10.1016/j.phrs.2010.01.014
Curl RL (1963) Dispersed phase mixing: I. Theory and effects in simple reactors. AIChE J 9(2):175–181. https://doi.org/10.1002/aic.690090207
D’Souza S (2014) A review of in vitro drug release test methods for nano-sized dosage forms. Adv Pharm 304757. https://doi.org/10.1155/2014/304757
Darminto, Cholishoh MN, Perdana FA, Baqiya MA, Mashuri, Cahyono Y, Triwikantoro (2011) Preparing Fe3O4 nanoparticles from Fe2+ ions source by co‐precipitation process in various pH. AIP Conference Proceedings 1415:234. https://doi.org/10.1063/1.3667264
De Jong WH, Borm PJA (2008) Drug delivery and nanoparticles:applications and hazards. Int J Nanomed 3(2):133–149. https://doi.org/10.2147/ijn.s596
Dong L, Yan L, Hou WG, Liu SJ (2010) Synthesis and release behavior of composites of camptothecin and layered double hydroxide. J Solid State Chem 183:1811–1816. https://doi.org/10.1016/j.jssc.2010.05.035
Dorniani D, Hussein MZB, Kura AU, Fakurazi S, Shaari AH, Ahmad Z (2012) Preparation of Fe3O4 magnetic nanoparticles coated with gallic acid for drug delivery. Int J Nanomed 7:5745
Dorniani D, Hussein M, Kura A, Fakurazi S, Shaari A, Ahmad Z (2013) Sustained release of prindopril erbumine from its chitosan-coated magnetic nanoparticles for biomedical applications. Int J Mol Sci 14:23639–23653. https://doi.org/10.3390/ijms141223639
Dorniani D, Kura AU, Hussein-Al-Ali SH, Hussein MZB, Fakurazi S, Shaari AH, Ahmad Z (2014) In vitro sustained release study of gallic acid coated with magnetite-PEG and magnetite-PVA for drug delivery system. Sci World J 416354. https://doi.org/10.1155/2014/416354
Faraji M, Yamini Y, Rezaee M (2010) Magnetic nanoparticles: synthesis, stabilization, functionalization, characterization, and applications. J Iran Chem Soc 7(1):1–37. https://doi.org/10.1007/BF03245856
Guerra D, Viana R, Airoldi C (2009) Adsorption of mercury cation on chemically modified clay. Mater Res Bull 44:485–491. https://doi.org/10.1016/j.materresbull.2008.08.002
Gunasundari E, Senthil Kumar P, Christopher FC, Arumugam T, Saravanan A (2017) Green synthesis of metal nanoparticles loaded ultrasonic-assisted Spirulina platensis using algal extract and their antimicrobial activity. IET Nanobiotechnol 11(6):754–758. https://doi.org/10.1049/iet-nbt.2016.0223
Javid A, Ahmadian S, Saboury AA, Kalantar SM, Rezaei-Zarchi S, Shahzad S (2014) Biocompatible APTES–PEG modified magnetite nanoparticles: effective carriers of antineoplastic agents to ovarian cancer. Appl Biochem Biotechnol 173(1):36–54. https://doi.org/10.1007/s12010-014-0740-6
Kayal S, Ramanujan R (2010) Doxorubicin loaded PVA coated iron oxide nanoparticles for targeted drug delivery. Mater Sci Eng C 30:484–490. https://doi.org/10.1016/j.msec.2010.01.006
Khan B, Nawaz M, Waseem M, Hussain R, Arif S, Price G, Haq S, Rehman W (2019) Adsorption of methylene blue onto size controlled magnetite nanoparticles. Mater Res Exp. https://doi.org/10.1088/2053-1591/ab2ef9
LaMer VK, Dinegar RH (1950) Theory, production and mechanism of formation of monodispersed hydrosols. J Am Chem Soc 72(11):4847–4854. https://doi.org/10.1021/ja01167a001
Mahmoudi M, Simchi A, Imani M, Milani AS, Stroeve P (2008) Optimal design and characterization of superparamagnetic iron oxide nanoparticles coated with polyvinyl alcohol for targeted delivery and imaging. J Phys Chem B 112(46):14470–14481. https://doi.org/10.1021/jp803016n
Mahmoudi M, Simchi A, Imani M (2009) Cytotoxicity of uncoated and polyvinyl alcohol coated superparamagnetic iron oxide nanoparticles. J Phys Chem C 113(22):9573–9580. https://doi.org/10.1021/jp9001516
Na K, Lee ES, Bae YH (2003) Adriamycin loaded pullulan acetate/sulfonamide conjugate nanoparticles responding to tumor pH: pH-dependent cell interaction, internalization and cytotoxicity in vitro. J Control Rel 87(1–3):3–13
Na K, Lee ES, Bae YH (2007) Self-organized nanogels responding to tumor extracellular pH: pH-dependent drug release and in vitro cytotoxicity against MCF-7 cells. Bioconj Chem 18(5):1568–1574
Niemann B, Sundmacher K (2010) Nanoparticle precipitation in microemulsions: population balance model and identification of bivariate droplet exchange kernel. J Coll Interf Sci 342(2):361–371. https://doi.org/10.1016/j.jcis.2009.10.066
Niemann B, Rauscher F, Adityawarman D, Voigt A, Sundmacher K (2006) Microemulsion-assisted precipitation of particles: experimental and model-based process analysis. Chem Eng Process Process Intensif 45(10):917–935. https://doi.org/10.1016/j.cep.2005.10.012
Niemann B, Veit P, Sundmacher K (2008) Nanoparticle precipitation in reverse microemulsions: particle formation dynamics and tailoring of particle size distributions. Langmuir 24(8):4320–4328. https://doi.org/10.1021/la703566v
Pang Y, Zeng G, Tang L, Zhang Y, Liu Y, Lei X, Li Z, Zhang J, Liu Z, Xiong Y (2011) Preparation and application of stability enhanced magnetic nanoparticles for rapid removal of Cr (VI). Chem Eng J 175:222–227
Paul KG, Frigo TB, Groman JY, Groman EV (2004) Synthesis of ultrasmall superparamagnetic iron oxides using reduced polysaccharides. Bioconj Chem 15(2):394–401. https://doi.org/10.1021/bc034194u
Plank C, Schillinger U, Scherer F, Bergemann C, Remy JS, Krötz F, Anton M, Lausier J, Rosenecker J (2003) The magnetofection method: using magnetic force to enhance gene delivery. Biol Chem 384:737–747. https://doi.org/10.1515/BC.2003.082
Prijic S, Sersa G (2011) Magnetic nanoparticles as targeted delivery systems in oncology. Radiol Oncol 45(1):1–16. https://doi.org/10.2478/v10019-011-0001-z
Saifuddin N, Wong CW, Nur Yasumira AA (2009) Rapid biosynthesis of silver nanoparticles using culture supernatant of bacteria with microwave irradiation. J Chem 6(1):61–70
Sun C, Sze R, Zhang M (2006a) Folic acid-PEG conjugated superparamagnetic nanoparticles for targeted cellular uptake and detection by MRI. J Biomed Mater Res A 78A(3):550–557. https://doi.org/10.1002/jbm.a.30781
Sun C, Sze R, Zhang M (2006b) Folic acid-PEG conjugated superparamagnetic nanoparticles for targeted cellular uptake and detection by MRI. J Biomed Mater Res A off J Soc Biomater Jpn Soc Biomater Aust Soc Biomater Korean Soc Biomater 78(3):550–557
Tan S, Wu T, Zhang D, Zhang Z (2015) Cell or cell membrane-based drug delivery systems. Theranostics 5(8):863–881. https://doi.org/10.7150/thno.11852
Tang M, Dou H, Sun K (2006) One-step synthesis of dextran-based stable nanoparticles assisted by self-assembly. Polymer 47:728–734. https://doi.org/10.1016/j.polymer.2005.11.091
Tao X, Jin S, Wu D, Ling K, Yuan L, Lin P, Xie Y, Yang X (2016) Effects of particle hydrophobicity, surface charge, media pH value and complexation with human serum albumin on drug release behavior of mitoxantrone-loaded pullulan nanoparticles. Nanomaterials 6(1):2
Thanh NTK, Maclean N, Mahiddine S (2014) Mechanisms of nucleation and growth of nanoparticles in solution. Chem Rev 114(15):7610–7630
Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J, Zboril R (2016) Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev 116(9):5338–5431
Vuković G, Marinković A, Obradović M, Radmilović V, Čolić M, Aleksić R, Uskoković PS (2009) Synthesis, characterization and cytotoxicity of surface amino-functionalized water-dispersible multi-walled carbon nanotubes. Appl Surf Sci 255(18):8067–8075. https://doi.org/10.1016/j.apsusc.2009.05.016
Zbigniew S, Budzyński M, Durak K, Czernel G (2017) Synthesis and characterization of iron oxide magnetic nanoparticles. Nukleonika 62(2):73–77
Zhang Y, Zhang L, Ban Q, Li J, Li C-H, Guan Y-Q (2018) Preparation and characterization of hydroxyapatite nanoparticles carrying insulin and gallic acid for insulin oral delivery. Nanomed Nanotechnol Biol Med 14(2):353–364
Acknowledgements
We are grateful to Dr. G. Kociok-Köhn for the XRD and to Dr. Rami for TG/DTA analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khan, B., Nawaz, M., Price, G.J. et al. In vitro sustained release of gallic acid from the size-controlled PEGylated magnetite nanoparticles. Chem. Pap. 75, 5339–5352 (2021). https://doi.org/10.1007/s11696-021-01724-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-021-01724-6