
Abstract
COD and TOC removal studies of mixture of two azo dyes viz. Ponceau Xylidine and calconcarboxylic acid by Fenton and Fenton-type processes were undertaken using \({\text{H}}_{2} {\text{O}}_{2}\) (HP) and \({\text{Na}}_{2} {\text{S}}_{2} {\text{O}}_{8}\) (SPS) as oxidants. A decolourisation of 94% in 1 min was achieved under optimised parameters, although mixing of dyes caused a retardation of mineralisation when compared with individual dyes. The study has established that treatment can be carried out at the natural pH of the dye solution in \({\text{Fe}}^{2 + }\)/SPS system. This makes the treatment process economically viable. \({\text{Fe}}^{2 + }\)/SPS system at pH 3 and 4.9 was found to be more efficient than \({\text{Fe}}^{2 + }\)/HP system as far as mineralisation is concerned. We could achieve 100% consumption of HP within 5 min of reaction, and therefore, no interference of HP on COD measurement is expected. The removal efficiency of different water quality parameters was found to be in the order: colour > \({\text{COD}}_{\text{eff}}\) > \({\text{TOC}}_{\text{eff}}\). Both COD and TOC removals followed pseudo-first-order kinetics. During the degradation process, 74% azo bonds were found to convert to \({\text{NH}}_{4}^{ + }\) and the rest to other nitrogenous products. Different ions and intermediates have been identified by ion chromatography. Based on the toxicity assay, we have recommended microorganism selection criteria for toxicity assay depending on the nature of recipient water body. Our results of detoxification measurement suggest that Escherichia coli-based assay could be used for fresh water and Vibrio fischeri for marine environment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Wastewater discharged from industries such as textile and leather industries is usually contaminated with high concentrations of organic chemicals. Such wastewater usually has high chemical oxygen demand (COD), total organic carbon (TOC) values and strong colour. Therefore, quantification of COD and TOC can potentially become indicators of organic pollutants in water. Conversely, the removal of COD and TOC form wastewater is an essential parameter for any treatment process. Advanced oxidation processes (AOPs), which produce \({\text{HO}}^{\cdot}\) and \({\text{SO}}_{4}^{ \cdot - }\) radicals, have been used as a popular tool by environmental chemists for the degradation of organic contaminants (Liu et al. 2018; Khan et al. 2017). The role of AOP in the degradation process is to reduce the organic molecules into low molecular weight compounds (COD and TOC reduction process) followed by oxidation of these compounds into \({\text{CO}}_{2}\), \({\text{H}}_{2} {\text{O}}\) and inorganic anions. Among the various AOPs, Fenton and Fenton-type processes (Eqs. 1 and 2) are convenient and cost-effective. Apart from these advantages, both the processes do not require energy for the activation of HP or SPS to generate \({\text{HO}}^{ \cdot}\) and \({\text{SO}}_{4}^{ \cdot - }\) radicals.
The wastewater is considered as non-biodegradable if the ratio of its biological oxygen demand (BOD) to COD, i.e. BOD/COD, is less than 0.4 (Aye et al. 2004; Tabrizi and Mehrvar 2004). Thus, determination of COD of wastewater at different stages of treatment assumes significance as it serves as an indicator of its biodegradability. There are reports where different AOPs have been used to reduce the COD value of wastewater with an aim to enhance its biodegradability (Ishak and Malakahmad 2013; Martins et al. 2010). Guo et al. (2018) were successful in achieving COD and TOC removal efficiency to the extent of 85.3 and 75.2%, respectively, when benzene dye intermediates were subjected to Fenton process. Electro-Fenton process was employed to reduce COD of rayon industry wastewater up to 88% (Ghosh et al. 2011). An increase in COD reduction was observed with an increase in HP concentration up to certain maximum, beyond which a decreasing trend was observed.
The literature dealing with degradation of single dyes is abundantly rich. Although dye and textile industries invariably use mixture of dyes, the decolourisation, mineralisation and COD removal studies of mixture of dyes are scarcely found in the literature. With an aim to enrich the knowledge on degradation of mixture of dyes, we have started with mixture of two azo dyes, viz. PX and CCA. Degradation and mineralisation studies of these two individual dyes by Fenton and Fenton-type processes have been reported from our laboratory (Marbaniang 2015; Sahoo et al. 2015). The present study was devoted to (1) find the most suitable operational parameters (e.g. concentration of dye in the mixture, \({\text{Fe}}^{2 + }\) and oxidant, and pH) for optimal degradation, COD and TOC removal efficiency (\({\text{COD}}_{\text{eff}}\) and \({\text{TOC}}_{\text{eff}}\), respectively) of the mixture of dyes by Fenton and Fenton-type processes using HP and SPS as oxidants, respectively; (2) determine the kinetics of COD and TOC removal; (3) identify the ions and intermediate products formed during the process; and (4) establish the mechanism of their formation. Since the presence of residual HP has been reported to influence the COD of a given solution (Kang et al. 1999), excess of its use is not recommended for the treatment process and hence it was important to determine the HP utilisation efficiency at the optimum parameters. The objective of any treatment process should not only be to remove COD and TOC, but it should be ensured that the resulting solutions after the treatment are not toxic to the aquatic microorganisms. Keeping this in mind, we have assessed the toxicity of the treated solutions during the treatment process and correlate them with mineralisation and COD at different stages of treatment. Environmental chemists generally adopt two different methods of toxicity assessment: (1) by measuring the growth inhibition assay (reduction of colony-forming unit) of Escherichia coli (E. coli); (2) by measuring inhibition of luminescence (light loss) by marine bacterium Vibrio fischeri (V. fischeri) when exposed to a test sample (Zhang et al. 2019a; Sahoo et al. 2018; Le et al. 2016; Munoz et al. 2012; Bayo et al. 2009). Vibrio fischeri emits light as a natural process of their metabolism. When exposed to a toxic chemical, a disruption of the respiratory process of the bacteria inhibits the emitted light intensity. In this study, we have compared the toxicity of the samples by these two methods at different stages of treatment in order to formulate microorganism selection criteria for toxicity assessment.
Materials and methods
Reagents
The azo dyes, Ponceau Xylidine (PX) (synonym: 1-(2,4-xylylazo)-2-naphthol-3,6-disulphonicacid disodium salt; molecular formula:\({\text{C}}_{18} {\text{H}}_{14} {\text{N}}_{2} {\text{Na}}_{2} {\text{O}}_{7} {\text{S}}_{2}\); molecular weight: 480.42 (g mol−1); \(\lambda_{\hbox{max} }\) = 504 nm; pH of the dye solution in water: 6.2) and calconcarboxylic acid (CCA) (synonym: 3-hydroxy-4-(2-hydroxy-4-sulfo-1-naphthylazo) naphthalene-2-carboxylic acid; molecular formula:\({\text{C}}_{21} {\text{H}}_{14} {\text{N}}_{2} {\text{O}}_{7} {\text{S}};\) molecular weight: 438.4146 (\({\text{g mol}}^{ - 1}\)); \(\lambda_{\hbox{max} }\) = 552 nm; pH of the dye solution in water: 4.8) were procured from Sigma-Aldrich (Germany). The molecular structures of PX and CCA are shown in Fig. 1.

Structures of Ponceau Xylidine (a) and calconcarboxylic acid (b)
The other chemicals, viz. sulphuric acid (\({\text{H}}_{2} {\text{SO}}_{4}\), GR), sodium hydroxide (NaOH, GR), hydrogen peroxide (\({\text{H}}_{2} {\text{O}}_{2}\), 30% w/w, purified), sodium peroxodisulphate (\({\text{Na}}_{2 } {\text{S}}_{2} {\text{O}}_{8}\), for analysis), iron (II) sulphate (\({\text{FeSO}}_{4} \cdot 7{\text{H}}_{2} {\text{O}}\), GR) were obtained from Merck. ‘Solution 1’ (mercuric sulphate and sulphuric acid) and ‘Solution 2’ for low range COD (silver sulphate, chromic acid, sulphuric acid and demineralised water) required for the determination of COD were supplied by HACH, USA. The chemicals used for ion chromatographic analyses were sodium hydroxide (NaOH, 50–52% in water, Sigma), methane sulphonic acid (\({\text{CH}}_{3} {\text{SO}}_{3} {\text{H}}\), HIMEDIA), acetonitrile (\({\text{CH}}_{3} {\text{CN}},\) HPLC grade, HIMEDIA), ammonium acetate (\({\text{CH}}_{3} {\text{COONH}}_{4}\), Merck, Emparta ACS). All the chemicals were used as received without further purification. Luria Bertoni (LB) agar, LB broth and ampicillin used in detoxification experiment were acquired from Himedia, India. The lyophilised luminescent bacteria V. fischeri used for toxicity assessment were supplied by Modern Water Inc, USA.
Procedure
Aqueous solution of PX and CCA of desired concentration was prepared by dissolving required amount in Millipore water (Elix3 Century, Millipore India, Bangalore). All the reactions were carried out in the presence of air and at room temperature (varying between 19 and 25 °C) by placing required volume of the dye solution of required concentration in amber borosilicate reagent bottles. It may be mentioned that the temperature was not controlled during course of the experiments. Each bottle was designated to be sacrificed for analysis after a predetermined time interval. The mouths of the bottles were covered with aluminium foil, and two holes were pierced through it to allow free passage of air. The initial pH of the solution was adjusted to the desired value with \({\text{H}}_{2} {\text{SO}}_{4}\) (the strength varying between 0.05 and 0.35 N depending on the pH required) or \({\text{NaOH}}\) (the strength varying between 0.025 and 0.125 N depending on the pH required) using a digital pH meter (Eutech instrument pH-Tutor). The reaction was initiated by adding 0.5 ml of each of desired metal ion solution of concentration 340 mM to the dye solution followed by adding equal volume of each of 1460 mM of each oxidant making the total volume 100 ml. Therefore, the actual strength of metal ions and oxidants in the solution was maintained at 1.7 mM and 7.3 mM, respectively. All the solutions used in this work were freshly prepared except the dye solution, which was stored at \(4\;^\circ {\text{C}}\) and used within 3 days. The reaction was continued without adjustment of pH, and any change (if any) during the course of the reaction was noted periodically.
Analytical techniques
Decolourisation studies were carried out by measuring the absorbance at 520 nm with the help of a UV–Vis spectrophotometer (HACH, USA; DR 6000). Mineralisation study was carried out by measuring the TOC of the samples using a TOC analyser (Make: Elementar Analysensysteme GmbH, Model: Liqui TOC II). All the analyses were done immediately after the reaction, and the reproducibility in TOC measurement was checked by frequent measurement of the standard (1:1 solution of potassium phthalate and sodium carbonate as TOC and total inorganic carbon standard, respectively) as suggested by the manufacturer and the error were always found to be within ± 5%.
The procedure for the determination of COD was supplied by HACH, USA (Eslami et al. 2013). In short, 2 ml of a given sample mixed with 0.25 ml of ‘Solution 1’ and 2.8 ml of ‘Solution 2’ (low range) was first digested in a COD digester (HACH, USA; DRB 200) at 150 °C for 2 h. The digested sample after cooling to room temperature was analysed at 420 nm with the help of a UV–Vis spectrophotometer (HACH, USA; DR 6000) for measurement of COD of the samples. The data presented in the text and figures were analysed by standard deviation using ‘Origin 7’ (Microcal Inc.) and have been rounded up to significant values.
Decolourisation, \({\text{COD}}_{\text{eff}}\) and \({\text{TOC}}_{\text{eff}}\) (mineralisation) are calculated according to the following equations:
where \(A_{0}\), \({\text{COD}}_{0}\) and \({\text{TOC}}_{0}\) are initial absorbance, COD and TOC, respectively, and \(A_{t}\), \({\text{COD}}_{\text{t}}\) and \({\text{TOC}}_{\text{t}}\) are absorbance, COD and TOC at time ‘t’, respectively.
The degradation rate constant in terms of COD removal was determined according to the pseudo-first-order rate laws (Eq. 6)
where \({\text{COD}}_{0}\) is the initial COD at time t = 0 and COD is its value at any time t (min). The first-order rate constant (\(k_{\text{COD}}\)) for COD removal is the slope of the straight line obtained by plotting \(- { \ln }({\text{COD}}/{\text{COD}}_{0} )\) vs. time. The kinetics of TOC removal was determined by replacing COD with TOC and \({\text{COD}}_{0}\) with \({\text{TOC}}_{0}\) in Eq. (6), where \({\text{TOC}}_{0}\) is the initial value of TOC at time t = 0 and TOC is its value at any time t (min).
Anions, cations, organic acids, phenols and concentration of residual HP were analysed using ion chromatography system (ICS) supplied by Thermo Scientific, USA (Dionex, ICS-1100). Anions and organic acids were analysed using an IonPac AS11 analytical column (4 × 250 mm) with a suitable guard column and an automatic electrolytic suppressor (AERS 500, 4 mm). The current of the suppressor was maintained at 30 mA for anions and 38 mA for organic acids. The mobile phase was NaOH (12 mM for anion and 15 mM for organic acids) with a flow rate of 1 ml min−1. Cations were analysed using IonPac CS17 analytical column (4 × 250 mm) with a suitable guard column and an automatic electrolytic suppressor (CERS 300, 4 mm) with a current of 15 mA. The mobile phase was 5 mM methane sulphonic acid (MSA) with a flow rate of 1 ml min−1. Concentration of residual HP was analysed using a CarboPac PA20 analytical column (3 × 150 mm) with a suitable guard column and an electro-chemical detector (ED 50A). The mobile phase was 50 mM NaOH with a flow rate of 0.5 ml min−1. Phenols were analysed using a VWD variable wavelength absorbance UV–Vis detector (cell path length: 10 mm; cell volume: 11 µL). The wavelengths selected for the purpose were 270 nm and 320 nm. The mobile phase was 0.1 M ammonium acetate solution with a flow rate of 1 ml min−1. It was prepared by mixing appropriate amount of ammonium acetate to a mixture of water and acetonitrile (1:1). All the experiments were repeated at least three times, and the error was always found to be within ± 5%. The data presented in the text and figures were analysed by standard deviation using ‘Origin 7’ (Microcal Inc.) and have been rounded up to significant values.
Toxicity assessment of the treated solutions
We have adopted two methods for the toxicity assessment of the treated solutions. The first method is based on the on E. coli growth inhibition (metabolic inhibition) bioassay. Several papers on biotoxicity assessment have been published in recent years (Zhang et al. 2019a; Sahoo et al. 2018). The procedure involves dissolving 40 g of LB agar in 1000 ml of water followed by autoclaving for 30 min. Upon cooling down to about 40 °C, ampicillin (1 µL for each ml of LB agar solution) was added and mixed, and aliquots of approximately 10 ml of the medium were poured into sterilised Petri plates. LB broth (25 g) was separately dissolved in 1000 ml of water, aliquoted (9 ml) in 100 ml conical flasks and sterilised by autoclaving for 30 min. To the sterilised LB broth, 10 µL of ampicillin, 100 µL of the E. coli culture inocula and 1 ml of the dye or other test solution were added, and the cultures were grown at 37 °C overnight in a rotary shaker. The following day, the mid-log phase culture was diluted 10,000 times and 10 µL of the diluted culture was spread over the LB agar plates prepared earlier. The plates were incubated for 16 h in a 37 °C incubator as described earlier (Sharan et al. 2007). Colony-forming units (CFUs) formed in each plate were counted. All the experiments were performed under sterilised conditions. The relative toxicity, measured as a reduction in CFU, of the treated solutions presented in the study was with respect to the control.
In the other method, the toxicity was measured using Microtox FX test system (Modern Water Inc, USA). This method is based on the principles suggested by ISO (2007) (Karczmarczyk et al. 2014). The relative toxicity of each sample was measured by using 81.9% screening test, the procedure of which was supplied by Modern Water Inc, USA. The optical density (OD) of the samples was checked to ensure that the absorbance is not greater than 1.0 at 490 nm. The test was conducted at 15–22 °C by adding osmotic adjusting solution (OAS) containing 22% NaCl to the samples to allow normal cellular activity of V. fischeri and emission of luminescence. The pH of the samples in the present case varies between 2.6 and 4.9 and therefore, was adjusted to ≈ 6 by using 5 N NaOH (for fine adjustment 0.5 N NaOH was used) before adding OAS. The samples are centrifuged at 4500 rpm for 3 min to remove the precipitate formed after the adjustment of pH. Microtox SOLO reagent (V. fischeri bacteria) was reconstituted by adding 300 µL Microtox diluent (3.5% NaCl) and was incubated for 15 min followed by the addition of samples containing OAS. The inhibition of luminescence or light loss (%) by the bacteria was analysed by the Microtox FX photometer. The light loss (%) was used as a measure of the relative toxicity of the samples.
Results and discussion
Decolourisation study and spectral analysis
A typical UV–Vis absorption spectra of the dye, PX, in its pure form exhibits peaks at 222, 332 and 504 nm and CCA at 232, 552 nm and a shoulder at 604 nm. However, the UV–Vis spectrum of mixture of dyes in equal proportions is characterised by three absorption bands—two appear in UV region (222 and 332 nm) and one in the visible region (520 nm) with a shoulder at 604 nm (Fig. 2). While the peaks at 222 and 232 nm in individual dyes merge together and appear at 222 nm, the peak at 332 nm remains unchanged in the mixture of dyes. At the same time, peaks at 504 and 552 nm appearing in PX and CCA, respectively, are absent in the mixture of dyes and are replaced by a new peak at 520 nm. In addition to this and due to obvious reason, there is an increase in the absorbance of mixture of dyes at both the peaks. Absorption peak at 222 and 332 nm (representing \(\pi \to \pi^{*}\) transition in aromatic rings) indicates the presence of aromatic rings in the mixture of the dyes. The band at 332 nm may also represent benzene or naphthalene rings attached to azo bond. The peak at 520 nm (representing \(n \to \pi^{*}\) transitions of N=N, C=N and C=O chromophore groups), responsible for the colour of the (PX + CCA) solution, is used to monitor the decolourisation of the solution by measuring the decrease in absorbance at 520 nm.

UV–Vis absorption spectra of PX, CCA and (PX + CCA): [PX] = [CCA] = 0.05 mM
It is established that the first step in the degradation of azo dyes involves the cleavage of –N=N– bond by the attack of \({\text{HO}}^{ \cdot}\) radicals generated in Fenton process (Eq. 1) (Panajkar and Mohan 1993). The cleavage of –N=N– causes a decrease in the intensity of colour of (PX = CCA) solution. The involvement of \({\text{HO}}^{ \cdot}\) radicals was established by carrying out the reaction in the presence tert-butanol, a known scavenger of \({\text{HO}}^{ \cdot}\) radicals. It was observed that the decolourisation reduced from 98.3 to 67.6% in 120 min in the presence of 0.1 mol l−1 of tert-butanol. The decolourisation further decreased with the increase in the concentration of the scavenger and reached a value of 44.7% at a concentration of 1.5 mol l−1. The UV–Vis absorption spectra of (PX + CCA) at different time of treatment are presented in Fig. 3. It is observed that the peak at 520 nm declines sharply in 5 min with a decolourisation of 94.1% and 100% in 120 min. On the other hand, the intensity of the peak at 222 nm remained constant throughout the reaction and that at 332 nm increased sharply within 5 min of reaction. This increase may be attributed to the accumulation of intermediate aromatic products. Similar observation was made in the degradation of mixture of dyes in Fenton-type process and hence is not presented here.

UV-Vis absorption spectra of mixture of dyes (PX + CCA) at different period of treatment: [PX] = [CCA] = 0.05 mM; [\({\text{Fe}}^{2 + }\)] = 1.7 mM; [HP] = 7.3 mM; pH = 3
In order to have a clear understanding on the decolourisation of mixture of dyes, we have compared the decolourisation of mixture of dyes (PX + CCA) with the individual dyes, viz. PX and CCA reported earlier from our laboratory (Table 1) (Marbaniang 2015; Sahoo et al. 2015). HP was used as oxidant in Fenton process of all the studies including the present study. While ammonium persulphate (APS) was used as oxidant in earlier studies involving PX and CCA in Fenton-type process, SPS was used in Fenton-type of the present study. The effect of mixing the dyes in the degradation process becomes quite apparent by looking at Table 1. The decolourisation was very fast in the beginning and (≈ 94% in both the treatment processes was completed within 1 min), beyond which the reaction proceeded extremely slowly and complete decolourisation was achieved in 120 min in Fenton process at pH 3 and 60 min in Fenton-type process at the natural pH of the dye solution, i.e. 4.9 (Fig. 4). Thus, one of the important effects of mixing the dyes is the possibility of carrying out the Fenton-type reaction of mixture of dyes at the natural pH (4.9 in the present study) of the dye mixture. Carrying out the reaction at this pH will be economically viable as no external reagent is required to be added to adjust the pH. Another important effect is the change of optimum pH of CCA from 1 to 3 in Fenton process and to 4.9 in Fenton-type process. The very fast rate of decolourisation may be due to the presence of excess soluble iron than what is required for the dye solution under consideration (Pouran et al. 2018).

COD, TOC and colour removal of mixture of dyes in different systems: [PX] = [CCA] = 0.05 mM; [\({\text{Fe}}^{2 + }\)] = 1.7 mM; [oxidant] = 7.3 mM
Optimisation of operational parameters
As stated, the mineralisation of two individual azo dyes, PX and CCA, under their optimal parameters was reported from our laboratory (Marbaniang 2015; Sahoo et al. 2015). In the present study we aimed to establish the effect of mixing these two dyes on their operational parameters, and TOC, COD and colour removal efficiencies were compared to their individual studies. The effect of colour removal has already been discussed in the previous section. The concentration of both the dyes in this study is the same as used in their individual studies. For Fenton process (\({\text{Fe}}^{2 + } /{\text{HP}}\)), concentration of \({\text{Fe}}^{2 + }\), HP and pH was optimised on the basis of \({\text{TOC}}_{\text{eff}}\). In Fenton-type process, only pH was optimised and the other parameters like concentration of \({\text{Fe}}^{2 + }\) and oxidant (SPS) were the same as those used in Fenton process.
The mineralisation of (PX + CCA) in Fenton process with \({\text{Fe}}^{2 + }\) dosage varying from 1.2 to 1.9 mM is presented in Fig. 5. A linear increase in mineralisation from 18 to 37.1% was observed when the dose was increased from 1.2 to 1.7 mM, beyond which a slight inhibition was observed. Thus 1.7 mM was considered as the optimum concentration of \({\text{Fe}}^{2 + }\). The inhibition effect on mineralisation may be due to the scavenging of \({\text{HO}}^{ \cdot}\) radicals by \({\text{Fe}}^{2 + }\) (Eq. 7) (Derbalah et al. 2004). Further, higher the concentration of \({\text{Fe}}^{2 + }\) higher the generation of \({\text{Fe}}^{3 + }\), which scavenges \({\text{HO}}^{ \cdot}\) radicals to form monohydroxy complex, \([{\text{FeOH}}]^{2 + }\) (Eq. 8).

Effect of [\({\text{Fe}}^{2 + } ]\) and pH (inset) on the mineralisation of (PX + CCA): [PX] = [CCA] = 0.05 mM; [oxidant] = 7.3 mM; treatment period = 120 min
The effect of pH on both \({\text{Fe}}^{2 + }\) /HP and \({\text{Fe}}^{2 + }\)/SPS systems was evaluated. In the former system, maximum mineralisation of 37.1% was achieved at pH 3, which progressively decreased with the increase in pH and reached the value of 20.6% at pH 11 (Inset of Fig. 5). Similar trend was also observed with decolourisation, which decreased from 100% at pH 3 to 63.8% at pH 11. The higher efficiency at pH 3 may be due to the higher oxidation potential of \({\text{HO}}^{ \cdot}\) radicals (Buxton et al. 1988) and the lower mineralisation at pH > 3 may be due to the oxidation of \({\text{Fe}}^{2 + }\) to \({\text{Fe}}\left( {\text{OH}} \right)_{3}\), which catalyses the decomposition of HP into \({\text{O}}_{2}\) and \({\text{H}}_{2} {\text{O}}\) (Badawy et al. 2006). Besides, oxidation of NaOH by HP (Eq. 9) and lower oxidation potential of \({\text{HO}}^{ \cdot}\) might have a contribution towards lower mineralisation at higher pH (Buxton et al. 1988). On the other hand, the mineralisation in \({\text{Fe}}^{2 + }\)/SPS system increases from 24.5% at pH 3 to 29.3% at pH 4.9 (natural pH of the pure dye solution), which decreases further on increasing pH and remains static (≈ 23%) at pH ≥ 7.
The effect of [HP] on the mineralisation was studied by carrying out the treatment process at different concentrations of HP keeping the other parameters constant. The mineralisation was found to increase from 23.2% at [HP] 6.7 mM to 37.1% at [HP] 7.3 mM. However, a low mineralisation of 33.9% was recorded when [HP] was increased to 7.5 mM. Therefore, 7.3 mM was regarded as the optimum [HP]. Since the mineralisation is due to the generation of \({\text{HO}}^{ \cdot}\) radicals in the \({\text{Fe}}^{2 + }\)/HP system, a higher degree of mineralisation is expected at higher [HP]. The result indicates that concentration of \({\text{HO}}^{ \cdot}\) radicals beyond a certain limit lowers mineralisation, and hence, maintaining right [HP] is essential to achieve a better treatment process. The decreases in the mineralisation at higher [HP] may be attributed to the scavenging of \({\text{HO}}^{ \cdot}\) radicals by HP (Eq. 10) to form hydroperoxyl radicals (\({\text{HO}}_{2}^{ \cdot}\)), which are less reactive and do not contribute to the degradation of organic molecules (Zhang et al. 2019b; Wan and Wang 2017). Simultaneous quenching of excess \({\text{HO}}^{ \cdot}\) radicals by \({\text{Fe}}^{2 + }\) (Eq. 7) is also another factor, which contributes to less availability of \({\text{HO}}^{ \cdot}\) radicals and hence reduces mineralisation (Zhang et al. 2019b). The consumption of HP as determined by ion chromatography is very fast, consuming 95.7% in 1 min, and the residual HP after 5 and 15 min of reaction was 0.8 and 0.2%, respectively. In other words, complete consumption of HP occurred within 5 min of reaction. Since the concentration of each of the two dyes in all the experiments described above is 0.05 mM, the optimum parameters of both the processes based on the above experiments are: [PX] = [CCA] = 0.05 mM; [\({\text{Fe}}^{2 + }\)] = 1.7 mM; [oxidant] = 7.3 mM, pH = 3 for \({\text{Fe}}^{2 + }\)/HP system and 4.9 for the other.
Since TOC is a measure of organic pollutants present in a wastewater sample, it is desired to achieve complete removal of TOC (mineralisation) in any treatment process. Under the present optimum parameters in \({\text{Fe}}^{2 + }\)/HP, \({\text{TOC}}_{\text{eff}}\) was found to be 37.1% in 120 min, which increased to 42.8% in 300 min (Table 2). On the other hand, the corresponding values in \({\text{Fe}}^{2 + }\)/SPS system were 57.8 and 56.3%, respectively, at pH 3 and 4.9. Thus, \({\text{Fe}}^{2 + }\)/SPS system is more efficient than \({\text{Fe}}^{2 + }\)/HP system at pH 3 and 4.9 as far as TOC removal is concerned. Another important effect of mixing the two dyes is the retardation of mineralisation when compared with individual dyes. While the values for PX and CCA in 240 min are 67.4 and 74.1%, respectively, in \({\text{Fe}}^{2 + }\)/HP system (Marbaniang 2015; Sahoo et al. 2015), that for the mixture of dyes is only 42.8% in 300 min at respective optimum parameters. Since TOC removal is lower than decolourisation, total colour removal does not provide any information on the purification and detoxification of a water sample.
COD removal study for \({\text{Fe}}^{2 + }\)/HP and \({\text{Fe}}^{2 + }\)/SPS systems
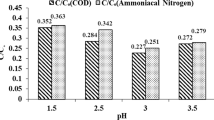
The \({\text{COD}}_{\text{eff}}\) in \({\text{Fe}}^{2 + }\) /HP system was studied at pH 3, whereas in \({\text{Fe}}^{2 + }\)/SPS system it was studied at pH 4.9 since COD removal is completely resisted at pH 3 (Table 2). A linear increase in \({\text{COD}}_{\text{eff}}\) from 52.5% in 5 min to 86.8% in 60 min was observed in the former system (Fig. 4). This is attributed to the degradation of dye solutions and the intermediates by \({\text{HO}}^{ \cdot}\) radicals. No change in \({\text{COD}}_{\text{eff}}\) was observed even after increasing the treatment period to 300 min. corresponding to a \({\text{TOC}}_{\text{eff}}\) value of 42.8%. COD removal, on the other hand, in \({\text{Fe}}^{2 + }\)/SPS system at pH 4.9 is resisted till 120 min with a \({\text{COD}}_{\text{eff}}\) value of only 13.1%. Thereafter, the increase was rather faster and reaches the value of 63.9% in 300 min, which corresponds to the \({\text{TOC}}_{\text{eff}}\) value of 56.3% (Fig. 4). The initial resistance of COD removal may be due to the presence of persulphate in the medium. From the above discussion, it is concluded that the removal efficiency of different water quality parameters is in the order: colour > \({\text{COD}}_{\text{eff}}\) > \({\text{TOC}}_{\text{eff}}\). The higher rate of colour removal than \({\text{COD}}_{\text{eff}}\) and \({\text{TOC}}_{\text{eff}}\) may be due to the generation of stable initial intermediate products, which require longer treatment period for further degradation by oxidation (Swaminathan et al. 2003). Since COD reflects the amount of oxidisable organic and inorganic species and TOC-only organic species, the fast rate of removal of COD than TOC may be attributed to the degradation of aromatic intermediates, resulting in the generation and accumulation of aliphatic organic acids like formic acid, malonic acid, etc. in the solution (Segura and Brillas 2017; Wan and Wang 2017). Based on this result, it may be recommended that TOC removal should form the basis of wastewater treatment. Since complete consumption of HP was observed within 5 min of reaction under the present conditions, the interference of HP on COD measurement is ruled out.
It is seen from Fig. 4 that the COD and TOC removal processes in both the systems consist of two stages: the first rapid process up to 60 min (1st stage) followed by slow degradation process (2nd stage). The rapid 1st stage is in consistent with the 95.7% consumption of HP in 1 min resulting in the COD removal of 52.5% in 5 min. and 86.8% in 60 min. The 2nd stage with negligible presence of HP resulted in no more COD removal in next 240 min. Similar trend was also observed with TOC removal process. While 33.3% mineralisation is obtained in 60 min (1st stage), only 9.5% in the next 240 min. (2nd stage). In other words, \({\text{COD}}_{\text{eff}}\) and \({\text{TOC}}_{\text{eff}}\) remain practically constant from 120 min onwards. The decrease in oxidation feasibility index (OFI), the ratio of COD to TOC, from the initial value of 4.8 to ≈ 1.0 in treatment period range of 60–300 min (Table 2) represents the attainment of more oxidisable state (Thomas et al. 2014). This is in accordance with the result that \({\text{COD}}_{\text{eff}}\) > \({\text{TOC}}_{\text{eff}}\). However, a reverse trend is observed for the COD removal in \({\text{Fe}}^{2 + }\)/SPS system, i.e. the slow 1st stage, where only 1.6% COD removal was obtained in 60 min and a rapid 2nd stage showing 62.3% COD removal in next 240 min. Contrary to the observation \({\text{COD}}_{\text{eff}}\) > \({\text{TOC}}_{\text{eff}}\) in \({\text{Fe}}^{2 + }\)/HP system, a reverse trend, i.e. \({\text{TOC}}_{\text{eff}}\) > \({\text{COD}}_{\text{eff}}\) was observed till 120 min of the reaction in \({\text{Fe}}^{2 + }\)/SPS system at pH 4.9. This result of initial stage is in accordance with the OFI values, which increased from the initial value of 4.8–5.9 in 120 min followed by a slow decrease up to 4.0 in 300 min. This result is in accordance with the increase in \({\text{COD}}_{\text{eff}}\) values from 120 min. Such observation was also reported earlier by Thomas et al. (2014) in their study on oxidative degradation of Acid Red 1 in aqueous medium.
Kinetics of COD and TOC removal
As discussed earlier, the COD and TOC removal follows a two-step process: first, oxidation of the dyes within 60 min of treatment followed by the slow oxidation of low molecular weight intermediates such as aliphatic organic compounds. Nevertheless, a deviation in COD removal in \({\text{Fe}}^{2 + }\)/SPS was observed. The pseudo-first-order kinetic plots of COD and TOC removal in different systems are presented in Figs. 6 and 7 and the corresponding rate constants are given in Table 3. The fact that no change in \({\text{COD}}_{\text{eff}}\) was observed in \({\text{Fe}}^{2 + }\) /HP system is shown by a slope of ‘0’ at pH 3. Data presented in Table 3 show that the rate constant of COD removal in the first stage is about 5 times higher than TOC removal in \({\text{Fe}}^{2 + }\) /HP system and TOC removal is about 20 times higher than COD removal at pH 4.9 in the initial fast process of \({\text{Fe}}^{2 + }\) /SPS system. Our result showing COD and TOC removal follows a pseudo-first-order kinetics is in good agreement with other studies involving degradation of pollutants (Abou-Elela et al. 2016; Bounab et al. 2015; Khieu et al. 2009). Further, a two-step pseudo-first-order kinetics as is described in the present study has been reported in previous studies also (Khieu et al. 2009; Herney-Ramirez et al. 2011).

TOC and COD removal kinetics in \({\text{Fe}}^{2 + }\)/HP system at pH 3

TOC and COD removal kinetics in \({\text{Fe}}^{2 + }\)/SPS system at pH 4.9
Analysis of ions and intermediate products
Identification of ions
Both PX and CCA have one source of nitrogen, i.e. two azo nitrogen atoms on each molecule. Under the present experimental conditions, \({\text{Na}}^{ + }\) and \({\text{SO}}_{4}^{2 - }\) are identified in both Fenton and Fenton-type processes. While \({\text{Na}}^{ + }\) is identified as the dissociation product of PX, \({\text{SO}}_{4}^{2 - }\) is formed as a result of substitution of \({\text{SO}}_{3}^{ - }\) groups present in both the dyes by \({\text{HO}}^{ \cdot}\) (Eqs. 11 and 12) (Karkmaz et al. 2004; Lachheb et al. 2002; Galindo et al. 2000). The \({\text{SO}}_{4}^{2 - }\) ion concentration determined was found to be higher than the theoretical values. This is due to the use of sulphuric acid to adjust the pH. The generation of \({\text{SO}}_{4}^{2 - }\) was observed within 5 min of reaction, during which time 94.1% decolourisation was achieved. So, it may be concluded that elimination of \({\text{SO}}_{4}^{2 - }\) and decolourisation takes place simultaneously.
The other inorganic ions identified are \({\text{NH}}_{4}^{ + }\) (in Fenton process) and \({\text{NO}}_{3}^{ - }\) (in Fenton-type process). The \({-}{\text{N}} = {\text{N}} -\) bonds of the azo dyes are attacked by \({\text{HO}}^{ \cdot}\), and transformed into amino groups, one on each aromatic ring. This provides a route for the generation of aromatic amino derivatives. The amino groups are then converted into \({\text{NH}}_{4}^{ + }\) by protonation of amino group followed by attack of \({\text{HO}}^{ \cdot}\) on the nitrogen-bearing carbon atom of the aromatic ring (Eqs. 13–15) (Sahel et al. 2007; Lachheb et al. 2002). This reaction may be accounted for the generation of phenol or its derivatives. The conversion of the azo group into \({\text{N}}_{2}\) gas has also been reported previously (Lachheb et al. 2002). The generation of \({\text{N}}_{2}\) may be due to the attack of \({\text{HO}}^{ \cdot}\) on the carbons attached to both ends of the \({-}{\text{N}} = {\text{N}} -\) bond (Eq. 16). This also provides an alternate route to the generation of aromatic hydroxyl derivatives. Considering the amount of \({\text{NH}}_{4}^{ + }\) formed in 300 min and the fact that no nitrate was observed in Fenton process, we can conclude that 74% of the azo bond is converted to \({\text{NH}}_{4}^{ + }\) and the rest 26% is may be accounted for other nitrogenous products including nitrogen gas. The conversion of azo bond to \({\text{NH}}_{4}^{ + }\) is shown in Fig. 8. It is observed that the conversion remains practically constant till 180 min of reaction varying from 32.4% in 5 min to 37.6% in 180 min. Thereafter, a sharp increase in ammonium ion formation to 73.8% in 300 min. was observed. Considering rapid decolourisation of 93.7% in 1 min and only 37.6% conversion of azo bond into ammonium ion in 180 min, it may be concluded that the azo bonds might have been broken completely to form aromatic amines (refer earlier discussion), but they could not be converted into ammonium ions and remain accumulated at aromatic amine stage in the final solution. This observation might explain the rise in intensity of (Fig. 3) absorption peak at 332 nm within 5 min of reaction. The increase in absorption of band at 332 nm, and therefore, the conclusion that aromatic intermediates are accumulated in the solution is finally corroborated by a low mineralisation of 42.7% in 300 min of Fenton reaction and 57.8 (pH 3) and 56.3% (pH 4.9) of Fenton-type reaction. Reddy and Mahajani (2005) have also suggested the conversion of \({\text{NH}}_{4}^{ + }\) to \({\text{NO}}_{3}^{ - }\) and \({\text{N}}_{2}\) gas (Eq. 17). Since no \({\text{NH}}_{4}^{ + }\) was detected in Fenton-type process, it may be concluded that \({\text{NH}}_{4}^{ + }\) ions are converted to \({\text{NO}}_{3}^{ - }\) as soon as they are formed. A list of ions and intermediates formed during the process as identified by ion chromatography is listed in Table 4.

Conversion of azo bond to \({\text{NH}}_{4}^{ + }\) with time: [PX] = [CCA] = 0.05 mM; [\({\text{Fe}}^{2 + }\)] = 1.7 mM; [HP] = 7.3 mM; pH = 3
Identification of intermediate products
Using the limited capability of ion chromatographic technique, we were able to identify phenol and its nitro and amino derivatives in Fenton-type and smaller aliphatic acids like malonic acid, formic acid and/or acetic acid (retention time for both are same), and maleic acid were identified in both the processes (Table 4). However, fumaric acid was identified in Fenton-type process only. The cleavage of azo dyes in the mixture (PX +CCA) proceeds with the attack of \({\text{HO}}^{ \cdot}\) to the \({-}{\text{N}} = {\text{N}} -\) chromophore with a probability of ~ 60% (Panajkar and Mohan 1993). There is evidence that successive addition of \({\text{HO}}^{ \cdot}\) to the \({\text{N}} = {\text{N}}\) bond generate derivatives of hydroxylamine which undergo further oxidation to produce nitroso followed by nitro aromatic compounds (Joseph et al. 2000). Substitution of nitro aromatic compounds by \({\text{HO}}^{ \cdot}\) produces hydroxybenzenes, which lead to the generation of aliphatic acids via the formation of quinones (Fendler and Gasowski 1968). Oxidation of the displaced \({\text{O}}_{2} {\text{N}}^{ \cdot}\) radical by \({\text{HO}}^{ \cdot}\) yields \({\text{HNO}}_{3}\) (Kotronarou et al. 1991). Based on the intermediate products and ions identified and backed by literature, rich on degradation mechanism of azo dyes (Steter et al. 2014; Neyens and Baeyens 2003; Joseph et al. 2000), the degradation mechanism of mixture of dyes (PX and CCA) is shown in Scheme 1.

Mechanism of degradation of mixture of dyes (PX + CCA)
Comparison of toxicity by CFU and luminescence inhibition assays
The relative toxicities of the mixture of pure dye as well as the solutions at different stages of treatment with \({\text{Fe}}^{2 + }\)/HP and \({\text{Fe}}^{2 + }\)/SPS systems were assessed as a function of CFU and inhibition of luminescence percentage of E. coli and V. fischeri, respectively. Both the methods did not show the same toxicity levels of a given sample. The relative toxicity of the solution of mixture of pure dyes was 20% more with V. fischeri-based assay than when measured with E. coli-based assay (Fig. 9) in \({\text{Fe}}^{2 + }\)/HP. When measured with the former, the relative toxicity fell to the extent of 50% in 5 min and was completely detoxified in 30 min. On the other hand, measurement with the later showed a slight increase (1%) in relative toxicity in 5 min probably due to the sensitivity of E. coli to some intermediate aromatic products formed in the initial stage of the treatment. A slow increase in detoxification was observed with treatment period, and complete detoxification was achieved in 300 min. The different levels of toxicity shown by the two methods may be attributed to the formation of different products generated at the initial and final stages of the treatment process. Since aromatic intermediates and organic acids are generated in the initial and final stages of the treatment, respectively, it may be concluded that E. coli is resistant to aromatic intermediates and V. fischeri to organic acids (Munoz et al. 2012).

Relative toxicity of treated solutions in \({\text{Fe}}^{2 + }\) /HP and \({\text{Fe}}^{2 + }\) /SPS systems: [PX] = [CCA] = 0.05 mM; pH of E. coli growth = 7; \([{\text{Fe}}^{2 + }\)] = 1.7 mM; [oxidant] = 7.3 mM
Although, the trend of relative toxicity in \({\text{Fe}}^{2 + }\)/SPS system is similar with \({\text{Fe}}^{2 + }\)/HP system, the solution remains toxic to both the species at the end of 300 min of treatment—5% more with E. coli than V. fischeri assays (inset of Fig. 9). One interesting feature in this system of treatment was the increase in toxicity in 5 min as shown by both the species. The inhibition of luminescence or light loss in pure dye solution was 64% which increased to 86% in 5 min of reaction. So, initially we assumed that the increased toxicity may be due to the presence of persulphate. To establish this, we have carried out the test with pure dye solution containing the same concentration of persulphate under similar operating parameters. In this case, the light loss was found to be 64% i.e., the same as that of pure dye. So, it is concluded that the increase in toxicity in 5 min may be due to the formation of toxic intermediate aromatic products. On further increasing the treatment period, the toxicity tends to decrease due to decomposition of intermediate products.
Since the sensitivities of methods to measure the toxicity were different with both the species, a selection of an assay should depend on the nature of the microorganisms present in the recipient water body where the detoxified wastewater is proposed to be discharged. Additional factors that can influence the selection of the method are (a) the cost involved and (b) the time factor. While measurement of toxicity with E. coli-based assay requires inexpensive and common instruments, a costly Microtox photometer is required for assay based on V. fischeri. On the other hand, while the toxicity measurement with V. fischeri-based assay requires only about 20 min, it takes about 3 days with E. coli-based assay. Therefore, we believe that measurement of toxicity with V. fischeri-based method could be a preferred choice when the detoxified wastewater recipient body is marine. However, when the recipient water body is fresh water, the method based on E. coli could be the recommended method for toxicity assessment of detoxified wastewater.
Conclusion
The study was devoted to evaluate the effect of mixing of two dyes (PX and CCA) on important water quality parameters like colour, COD and TOC. When compared with the result with individual dyes, four important effects are observed: (1) rapid decolourisation to the extent of ≈ 94% in 1 min and reduction in time for complete decolourisation by more than half in Fenton process and 75% in Fenton-type; (2) change of optimum pH of CCA from 1 to 3 in Fenton process and to 4.9 in Fenton-type process; (3) possibility of carrying out treatment at the natural pH of the dye solution; (4) reduction in magnitude of mineralisation. Of the two processes studied, \({\text{Fe}}^{2 + }\)/SPS system at pH 3 and 4.9 is more efficient than \({\text{Fe}}^{2 + }\)/HP system as far as mineralisation is concerned. Nevertheless, the COD removal efficiency is higher in \({\text{Fe}}^{2 + }\)/HP than the other. Since complete consumption of HP is observed in 5 min of the reaction, its effect on COD estimation is negated. The higher degradation at the natural pH of the dye solution (pH 4.9) in \({\text{Fe}}^{2 + }\)/SPS system implies that the process is economically viable as no external acid or base is required to be added to adjust the pH. The rate of removal of different parameters follows the order: colour > \({\text{COD}}_{\text{eff}}\) > \({\text{TOC}}_{\text{eff}}\) in \({\text{Fe}}^{2 + }\)/HP. However, the order in the 2nd and 3rd parameter within 120 min is reversed in \({\text{Fe}}^{2 + }\)/SPS system. Different ions such as \({\text{Na}}^{ + }\), \({\text{NH}}_{4}^{ + }\), \({\text{SO}}_{4}^{2 - }\), \({\text{NO}}_{3}^{ - }\) and intermediates such as phenol, formic acid, malonic acid, maleic acid and fumaric acid are identified by ion chromatography. Kinetic data of COD and TOC removal studies show that the rate constant of COD removal in the first stage is about 5 times higher than TOC removal in \({\text{Fe}}^{2 + }\)/HP system and TOC removal is about 20 times higher than COD removal at pH 4.9 in the initial fast process of \({\text{Fe}}^{2 + }\)/SPS system. The conversion azo bond to \({\text{NH}}_{4}^{ + }\) is an important part of degradation process, and it was estimated that 74% of azo bonds are converted to \({\text{NH}}_{4}^{ + }\) and the rest to other nitrogenous products including nitrogen gas. Detoxification of the treated solutions should be one of the important water quality parameters, which should be ensured before being released into water bodies. A comparison of toxicity assay of the treated solutions by E. coli and V. fischeri shows that both the bacteria display different sensitivity to a given test solution, which is evident from the fact that the relative toxicity of the solution of mixture of dyes is 20% more with V. fischeri than when measured with E. coli. Interestingly, complete detoxification was shown in 30 min by V. fischeri and 300 min by E. coli assay. Since V. fischeri is a marine bacterium, the assay with it should be performed in marine environment.
Abbreviations
- °C:
-
Degree Celcius
- g:
-
Gram
- h:
-
Hour
- hrs:
-
Hours
- L:
-
Litre
- min:
-
Minute
- mA:
-
Milliampere
- mm:
-
Millimetre
- mM:
-
Millimole
- ml:
-
Millilitre
- M:
-
Mole
- mol:
-
Mole
- nm:
-
Nanometre
- %:
-
Percentage
- N:
-
Normal
- W/W:
-
Weight/weight
- λ :
-
Lambda
- \(\mu\) :
-
Mu
References
Abou-Elela SI, Ali MEM, Ibrahim HS (2016) Combined treatment of retting flax wastewater using Fenton oxidation and granular activated carbon. Arab J Chem 9(4):511–517. https://doi.org/10.1016/j.arabjc.2014.01.010
Aye T, Mehrvar M, Anderson WA (2004) Effects of photocatalysis on the biodegradability of Cibacron Brilliant Yellow 3G-P (Reactive Yellow 2). J Environ Sci Heal A 39:113–126. https://doi.org/10.1081/ESE-120027372
Badawy MI, Ghaly MY, Gad-Allah TA (2006) Advanced oxidation processes for the removal of organophosphorus pesticides from wastewater. Desalination 194(1–3):166–175. https://doi.org/10.1016/j.desal.2005.09.027
Bayo J, Angosto JM, Gómez-López MD (2009) Ecotoxicological screening of reclaimed disinfected wastewater by Vibrio fischeri bioassay after a chlorination–dechlorination process. J Hazard Mater 172:166–171. https://doi.org/10.1016/j.jhazmat.2009.06.157
Bounab L, Iglesias O, Gonzalez-Romero E, Pazosb M, Sanroman MA (2015) Effective heterogeneous electro-Fenton process of m-cresol with iron loaded actived carbon. RSC Adv 5:31049–31056. https://doi.org/10.1039/c5ra03050a
Buxton GV, Greenstock CL, Helman WP, Ross AB (1988) Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH·/O·−) in aqueous solution. J Phys Chem Ref Data 17:513–886. https://doi.org/10.1063/1.555805
Derbalah AS, Nakatani N, Sakugawa H (2004) Photocatalytic removal of fenitrothion in pure and natural waters by photo-Fenton reaction. Chemosphere 57:635–644. https://doi.org/10.1016/j.chemosphere.2004.08.025
Eslami A, Moradi M, Ghanbari F, Mehdipour F (2013) Decolorization and COD removal from real textile wastewater by chemical and electrochemical Fenton processes: a comparative study. J Environ Health Sci Eng 11:1–8. https://doi.org/10.1186/2052-336X-11-31
Fendler JH, Gasowski GL (1968) Radiation-Induced hydroxylation of nitrobenzene in dilute aqueous solution. J Org Chem 33(5):1865–1868. https://doi.org/10.1021/jo01269a035
Galindo C, Jacques P, Kalt A (2000) Photodegradation of the aminoazobenzene acid orange 52 by three advanced oxidation processes: UV/H2O2, UV/TiO2 and Vis/TiO2: comparative mechanistic and kinetic investigations. J Photochem Photobiol A: Chem 130:35–47. https://doi.org/10.1016/S1010-6030(99)00199-9
Ghosh P, Samanta AN, Ray S (2011) Reduction of COD and removal of Zn2+ from rayon industry wastewater by combined electro-Fenton treatment and chemical precipitation. Desalination 266(1–3):213–217. https://doi.org/10.1016/j.desal.2010.08.029
Guo Y, Xue Q, Zhang H, Wang N, Chang S, Wang H, Pang H, Chen H (2018) Treatment of real benzene dye intermediates wastewater by the Fenton method: characteristics and multi-response optimization. RSC Adv 8(1):80–90. https://doi.org/10.1039/C7RA09404C
Herney-Ramirez J, Silva AMT, Vicente MA, Costa CA, Madeiraa LM (2011) Degradation of Acid Orange 7 using a saponite-based catalyst in wet hydrogen peroxide oxidation: kinetic study with the Fermi’s equation. Appl Catal B Environ 101(3–4):197–205. https://doi.org/10.1016/j.apcatb.2010.09.020
Ishak S, Malakahmad A (2013) Optimization of Fenton process for refinery wastewater biodegradability augmentation. Korean J Chem Eng 30(5):1083–1090. https://doi.org/10.1007/s11814-013-0002-2
Joseph JM, Destaillats H, Hung H-M, Hoffmann MR (2000) The sonochemical degradation of azobenzene and related azo dyes: rate enhancements via Fenton’s reactions. J Phys Chem A 104(2):301–307. https://doi.org/10.1021/jp992354m
Kang YW, Cho M-J, Hwang K-Y (1999) Correction of hydrogen peroxide interference on standard chemical oxygen demand test. Water Res 33(5):1247–1251. https://doi.org/10.1016/S0043-1354(98)00315-7
Karczmarczyk A, Celebanska A, Nogala W, Sashuk V, Chernyaeva O, Opallo M (2014) Electrocatalytic glucose oxidation at gold and gold-carbon nanoparticulate film prepared from oppositely charged nanoparticles. Electrochim Acta 117:211–216. https://doi.org/10.1016/j.electacta.2013.11.049
Karkmaz M, Puzenat E, Guillard C, Herrmann JM (2004) Photocatalytic degradation of the alimentary azo dye amaranth mineralization of the azo group to nitrogen. Appl Catal B Environ 51(3):183–194. https://doi.org/10.1016/j.apcatb.2004.02.009
Khan JA, He X, Shah NS, Sayed M, Khan HM, Dionysiou DD (2017) Degradation kinetics and mechanism of desethyl-atrazine and desisopropyl-atrazine in water with HO· and SO·−4 based-AOPs. Chem Eng J 325:485–494. https://doi.org/10.1016/j.cej.2017.05.011
Khieu DQ, Quang DT, Lam TD, Phu NH, Lee JH, Kim JS (2009) Fe-MCM-41 with highly ordered mesoporous structure and high Fe content: synthesis and application in heterogeneous catalytic wet oxidation of phenol. J Incl Phenom Macrocycl Chem 65:73–81. https://doi.org/10.1007/s10847-009-9624-8
Kotronarou A, Mills G, Hoffmann MR (1991) Ultrasonic irradiation of p-nitrophenol in aqueous solution. J Phys Chem 95:3630–3638. https://doi.org/10.1021/j100162a037
Lachheb H, Puzenat E, Houas A, Ksibi M, Elaloui E, Guillard C, Herrmann J (2002) Photocatalytic degradation of various types of dyes (Alizarin S, Crocein Orange G, Methyl Red, Congo Red, Methylene Blue) in water by UV-irradiated titania. Appl Catal B Environ 39:75–90. https://doi.org/10.1016/S0926-3373(02)00078-4
Le TXH, Nguyen TV, Yacouba ZA, Zoungrana L, Avril F, Petit E, Mendret J, Bonniol V, Bechelany M, Lacour S, Lesage G, Cretin M (2016) Toxicity removal assessments related to degradation pathways of azo dyes: toward an optimization of Electro-Fenton treatment. Chemosphere 161:308–318. https://doi.org/10.1016/j.chemosphere.2016.06.108
Liu Y, Fan Q, Wang J (2018) Zn-Fe-CNTs catalytic in situ generation of H2O2 for Fenton-like degradation of sulfamethoxazole. J Hazard Mater 342:166–176. https://doi.org/10.1016/j.jhazmat.2017.08.016
Marbaniang M (2015) Comparative studies on decolorization and mineralization of Ponceau S and Ponceau Xylidine by some advanced oxidation processes. Ph.D thesis, North Eastern Hill University, Shillong
Martins RC, Rossi AF, Quinta-Ferreira RM (2010) Fenton’s oxidation process for phenolic wastewater remediation and biodegradability enhancement. J Hazard Mater 180(1–3):716–721. https://doi.org/10.1016/j.jhazmat.2010.04.098
Munoz M, de Pedro ZM, Casas JA, Rodriguez JJ (2012) Triclosan breakdown by Fenton-like oxidation. Chem Eng J 198–199:275–281. https://doi.org/10.1016/j.cej.2012.05.097
Neyens E, Baeyens J (2003) A review of classic Fenton’s peroxidation as an advanced oxidation technique. J Hazard Mater B 98(1–3):33–50. https://doi.org/10.1016/S0304-3894(02)00282-0
Panajkar MS, Mohan H (1993) Investigations of transients produced on reactions of radicals with azobenzenes in aqueous solutions. Indian J Chem 32:25–27
Pouran SR, Bayrami A, Shafeeyan MS, Raman AAA, Daud WM (2018) A Comparative study on a cationic dye removal through homogeneous and heterogeneous fenton oxidation systems. Acta Chim Slov 65:166–171. https://doi.org/10.17344/acsi.2017.3732
Reddy GR, Mahajani VV (2005) Insight into wet oxidation of aqueous aniline over a Ru/SiO2 catalyst. Ind Eng Chem Res 44(19):7320–7328. https://doi.org/10.1021/ie050438d
Sahel K, Perol N, Chermette H, Bordes C, Derriche Z, Guillard C (2007) Photocatalytic decolorization of Remazol Black 5 (RB5) and Procion Red MX-5B-Isotherm of adsorption, kinetic of decolorization and mineralization. Appl Catal B Environ 77(1–2):100–109. https://doi.org/10.1016/j.apcatb.2007.06.016
Sahoo MK, Sinha B, Sharan RN (2015) Metal ion-catalyzed mineralization and biodetoxification studies of Calconcarboxylic acid in aqueous solution: effect of –COOH group. Desalin Water Treat 56(7):1955–1963. https://doi.org/10.1080/19443994.2014.956342
Sahoo MK, Kumar JE, Sinha B, Marbaniang M, Sharan RN (2018) Comparative studies of mineralization and detoxification of Mordant Black 17 in aqueous solution by UV light induced Mn+/H2O2 and Mn+/S2O82− systems. Water Sci Technol 77(12):2917–2928. https://doi.org/10.2166/wst.2018.290
Segura SG, Brillas E (2017) Applied photoelectrocatalysis on the degradation of organic pollutants in wastewaters. J Photoch Photobio B 31:1–35. https://doi.org/10.1016/j.jphotochemrev.2017.01.005
Sharan RN, Ryo H, Nomura T (2007) Critical role of RecA and RecF proteins in strand break rejoining and maintenance of fidelity of rejoining following γ-radiation induced damage to pMTa4 DNA in E. coli. Int J Radiat Biol 83(2):89–97. https://doi.org/10.1080/09553000601121140
Steter JR, Barros WRP, Lanza MRV, Motheo AJ (2014) Electrochemical and sonoelectrochemical processes applied to amaranth dye degradation. Chemosphere 117:200–207. https://doi.org/10.1016/j.chemosphere.2014.06.085
Swaminathan K, Sandhya S, Sophia AC, Pachhade K, Subrahmanyam YV (2003) Decolorization and degradation of H-acid and other dyes using ferrous–hydrogen peroxide system. Chemosphere 50:619–625. https://doi.org/10.1016/S0045-6535(02)00615-X
Tabrizi GB, Mehrvar M (2004) Integration of advanced oxidation technologies and biological processes: recent developments, trends, and advances. J Environ Sci Heal A 39(11–12):3029–3081. https://doi.org/10.1081/ESE-200034939
Thomas S, Sreekanth R, Sijumon VA, Aravind UK, Aravindakumar CT (2014) Oxidative degradation of Acid Red 1 in aqueous medium. Chem Eng J 244:473–482. https://doi.org/10.1016/j.cej.2014.01.037
Wan Z, Wang J (2017) Fenton-like degradation of sulfamethazine using Fe3O4/Mn3O4 nanocomposite catalyst: kinetics and catalytic mechanism. Environ Sci Pollut Res 24:568–577. https://doi.org/10.1007/s11356-016-7768-9
Zhang Y, Zhang N, Wang T, Huang H, Chen Y, Li Z, Zou Z (2019a) Heterogeneous degradation of organic contaminants in the photo-Fenton reaction employing pure cubic β-Fe2O3. Appl Catal B Environ 245:410–419. https://doi.org/10.1016/j.apcatb.2019.01.003
Zhang M, Dong H, Zhao L, Wang D, Meng D (2019b) A review on Fenton process for organic wastewater treatment based on optimization perspective. Sci Total Environ 670:110–121. https://doi.org/10.1016/j.scitotenv.2019.03.180
Acknowledgements
The authors gratefully acknowledge use of facilities acquired through the DAE-BRNS grant (2013/36/50-BRNS/2485, dated 05.12.2013) to MKS; DST-FIST grant (SR/FST/CSI-194-2008) of the Department of Science and Technology, Govt. of India, and UGC-SAP CAS-I grant (F.540/21/CAS/2013(SAP I)) of UGC to the Department of Chemistry, North-Eastern Hill University (NEHU), Shillong. Discussion with Prof. S. Mitra and Dr. D. P. S. Negi of Department of Chemistry, North-Eastern Hill University (NEHU), Shillong, on the kinetic study is duly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kumar, J.E., Mulai, T., Kharmawphlang, W. et al. Decolourisation, mineralisation and detoxification of mixture of azo dyes using Fenton and Fenton-type advanced oxidation processes. Chem. Pap. 74, 3145–3159 (2020). https://doi.org/10.1007/s11696-020-01147-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01147-9