
Abstract
Due to the lack of the acid 1,4-α-glucosidase, the urine of Pompe patients contains increased levels of specific glucose tetrasaccharide αGlc(1 → 6)αGlc(1 → 4)αGlc(1 → 4)Glc (Glc4), which was approved as a non-invasive biomarker of glycogen accumulation in the muscles of Pompe patients. MALDI-TOF mass spectrometry and 1H NMR spectroscopy were used for Glc4 detection in the urine samples of four Pompe patients with infantile, juvenile, and adult forms of the disease, before and during, or before and after, enzyme replacement therapy. The increased sensitivity and decreased urine volume required for Glc4 detection by MALDI-TOF mass spectrometry was obtained by specific derivatization directly on the MALDI plate by 3-aminoquinoline. This method is a suitable and fast method for Pompe screening. 1H NMR spectra, used for precise Glc4 quantifications, were proposed for the enzyme replacement therapy efficiency monitoring. Besides the clinical features and the biochemical parameters, knowledge about the evolution of the Glc4 levels over time during the administration of the therapy could help monitoring of the patient outcome as part of the standard protocol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pompe disease (OMIM#232300), also known as glycogen storage disease type II (GSD type II), is caused by a deficiency in, or absence of, the lysosomal enzyme acid 1,4-α-glucosidase (GAA, EC 3.2.1.20), which participate in glycogen degradation (Lim et al. 2014). As a consequence, glycogen accumulates in the tissues of patients, resulting in an altered neuromuscular structure and function. Depending on the patient’s age at symptoms onset, the Pompe disease is classified as infantile (IOPD) when it appears in the first year, or late onset (LOPD; childhood, juvenile, and adult form) (Dasouki et al. 2014). Pompe disease belongs to the oligosaccharidoses, a subgroup of lysosomal storage diseases. Due to the wide variety of the age onset, the non-specific clinical symptoms, and phenotypes, it is difficult to diagnose. Some cases of Pompe disease, especially those with late onset, have been misdiagnosed. Thus, diagnosis of Pompe disease is challenging and often significantly delayed, which increases the morbidity and early mortality of the disease (van der Ploeg and Reuser 2008; Pérez-López et al. 2015). Of the multiple methods applied for Pompe diagnosis, the GAA enzyme deficiency demonstration is the most often used (Kishnani et al. 2014). However, screenings based on GAA activity monitoring diagnosed Pompe positive patients, from which a majority were later revealed as false positives after a mutation analysis (Schielen et al. 2017). The whole diagnostic process may thus become time consuming and critical, particularly for patients with the most severe infantile form of Pompe.
Hallgren et al. 1974 found that the urine of patients with Pompe disease contains higher quantities of limiting glucose tetrasaccharide αGlc(1 → 6)αGlc(1 → 4)αGlc(1 → 4)Glc (Glc4), created during glycogen degradation. Elevated Glc4 levels were found in the urine of patients with other disorders (e.g., GSD III and VI, Duchene muscular dystrophy), and thus, it cannot serve as a general specific Pompe biomarker. Nevertheless, studies have shown that Glc4 quantities correlate with the glycogen content in the muscles of patients with the IOPD form of Pompe, and thus, it was proposed as a non-invasive biomarker of glycogen accumulation, particularly for the diagnosis of the IOPD Pompe form (Young et al. 2012; Manwaring et al. 2012; de Almeida et al. 2017).
In biochemical screening for oligosaccharidoses, the most widely used, but time consuming, thin-layer chromatography (TLC) has been gradually replaced with diverse analytical technologies for the detection of oligosaccharides accumulated in the urine. New high-throughput technologies challenge researchers to invent new, fast, and more reliable methodologies (Casado et al. 2017). In this review, they described different, frequently used techniques for the diagnostics of oligosaccharidoses. Most of them require sample pre-treatment (e.g., deproteinization, derivatization, and purification) to increase the sensitivity. However, these additional procedures modify a complex matrix of the body fluid and might increase the degree of inaccuracy of the results obtained. Furthermore, many techniques are not able to distinguish Glc4 isomers and can determine only the total Hex4 content. However, Young et al. 2012 concluded that Glc4 accounts for at least 95% of the total Hex4 fraction in the urine.
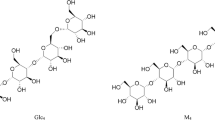
High-performance liquid chromatography (HPLC) methodology (Manwaring et al. 2012; Lobato et al. 2013), often in conjunction with mass spectrometry (An et al. 2000, 2005), is frequently used for Glc4 quantification in Pompe patients. As Glc4 is also present in healthy subjects and its quantity is age dependent, characteristic values for patients, both ill and healthy, were also searched (An et al. 2000; Young et al. 2012; Sluiter et al. 2012; Lobato et al. 2013). For mass spectrometric analyses, saccharides need to be derivatized to increase the sensitivity and intensities of the signals (Casado et al. 2017). Traditional derivatization methods usually use purification steps, causing a sample loss (Kaneshiro et al. 2011) and leading to inaccuracies in Glc4 quantification. Electrospray ionization (ESI) analysis of derivatized body fluids showed that samples from Pompe patients contain elevated levels of Glc4 and higher oligosaccharides [composed of up to seven hexose sugar units (Hex7)] in comparison with healthy subjects (Rozaklis et al. 2002). In 2012, Sluiter et al. 2012 developed a fast UPLC-MS/MS assay, selective for Pompe Glc4 detection, without the need for sample derivatization, and without interfering with the M4 maltodextrin tetrasaccharide ([→ 4)αGlc(1 →]4). Their study calculated age-matched Glc4 concentration values in Pompe patients (n = 66, IOPD and LOPD) and control individuals (n = 116). Large scale newborn screening performed by stable isotope dilution ESI–MS/MS revealed age-matched median values and ranges for baseline Glc4 levels in patients with IOPD, LOPD, and a pseudo-deficiency group of patients (Chien et al. 2015a, b). This study also estimated reference Glc4 levels for the control group of healthy babies aged 6 months or younger. The LC–MS/MS study of Young et al. 2012 quantified Glc4 in a large cohort of 208 Pompe disease patients and showed the age-matched upper control limits. Their study showed that urinary Glc4 is a biomarker of glycogen storage with 95% sensitivity and 84% specificity. They also confirmed the utility of Glc4 as a predictive biomarker of glycogen storage, reflecting the degree of disease severity in Pompe patients.
Due to its non-quantitative nature, the MALDI methodology (Szájli et al. 2008) can be used only for Hex4 detection, but not for its quantification. In 2013, Xia et al. 2013 revealed the characteristic oligosaccharide patterns of 11 different diseases by MALDI-TOF MS analysis of per-methylated samples of urine. One drawback of this approach was a laborious multistep derivatization procedure used prior to the measurement. Bonesso et al. 2014 developed a simpler MALDI-TOF MS method for obtaining MS/MS oligosaccharide signatures. This rapid method, using 2,5-di-hydroxybenzoic acid (DHB) and 3-aminoquinoline (3-AQ) matrices, was shown to be suitable for the screening and identification of eight oligosaccharidoses. However, 3-AQ was used as a traditional MALDI matrix, and its potential to bind to reducing ends of oligosaccharides, to increase selectivity, was not revealed in this study.
Capillary electrophoresis (CE-LIF)-based procedures were developed and validated for the diagnostics of different oligosaccharidoses and related disorders using time-consuming sample derivatization with 8-aminopyrene-1,3,6-trisulphonate (APTS) before LIF detection (Casado et al. 2014).
NMR spectroscopy belongs, together with mass spectrometry, to the most frequently used high-throughput technologies in metabolomics studies. Detailed structural analysis of α-limit dextrins, oligosaccharides issued from glycogen degradation, and which were isolated from the urine of patients suffering from GSD type II and III (including GC–MS, direct probe MS and NMR) was performed by Kumlien et al. 1989. The application of 1H NMR and MALDI-TOF MS in Pompe disease screening was demonstrated by Pakanová et al. 2016. In that study, the DHB matrix was used for MALDI-TOF MS and LIFT MS/MS measurements. For Glc4 identification from 1H NMR spectra, the isolated H1 anomeric signal of a terminal 1,6-linked αGlc unit was used and proposed for its quantification.
In 2006, the FDA approved the first specific treatment for muscle disorder by recombinant human alglucosidase alpha (Myozyme®, Genzyme Corporation, USA), which was the first enzymatic replacement therapy (ERT) of Pompe disease (Burton. 2012). Published results have proven that the therapy prolongs the life of patients, improves motor functions in the infantile form of the disease (Kishnani et al. 2007, 2014; Amalfitano et al. 2001), normalises heart size, muscle pathology, and growth, and motoric development becomes adequate to age (Chien et al. 2009). In 2009, the FDA approved a second form of recombinant human alglucosidase alpha (Lumizyme®, Genzyme Corporation, USA) for the treatment of patients older than 8 years with late onset of Pompe disease, without signs of cardiac hypertrophy. Clinical tests showed a stabilization of neuromuscular deficits (Strothotte et al. 2010) after 3 months of therapy and the GAA activity in the skeletal muscles was corrected to 100% (van den Hout et al. 2004). Long-term administration (≈ 2 to 3 years) of ERT in adult Pompe patients positively affects muscle strength, pulmonary function, and daily life activities (Kuperus et al. 2017. In symptomatic patients, only an early diagnostics with early onset of the therapy can avoid irreversible damage to the organism (van den Hout et al. 2004). Patients who started ERT at < 3 months of age had better survival rates at 36 months than patients who started ERT at ≥ 3 months of age (Chien et al. 2015). In patients undergoing ERT, the monitoring of Glc4 should become a part of standard diagnostic protocols. Monitoring the Glc4 level in the urine of Pompe patients, as a response to ERT, was proposed by An et al. 2000 and Young et al. 2009, 2012.
Some countries screen newborns for lysosomal storage diseases. A long-term study dedicated to newborn screening was published by the team of Huang et al. (2015). They used the Glc4 level to distinguish newborns with late Pompe onset (LOPD) from newborns with low GAA activity caused by a pseudo-deficiency of the GAA allele (G576S). The LOPD patients had very high levels of Glc4 before ERT in comparison with those with a pseudo-deficient GAA allele (G576S), for which the Glc4 level was normal. They also found that the Glc4 level correlated with the serum creatinine kinase (CK) level in Pompe patients, confirming that Glc4 is an appropriate biomarker for IOPD newborns, who can thus receive ERT shortly after birth.
Multiple reasons support the importance of Glc4 quantity determination: (i) screening for individuals suspected of having the IOPD form, when the damage to the organism is progressing very fast and the patient would otherwise die before the age of 1 year; (ii) to discriminate between Pompe patients and those with the GAA pseudo-deficiency; and (iii) to monitor the efficiency of the ERT.
During recent decades, traditional thin-layer chromatography was used for the detection of Glc4, as a complementary method to the estimation of GAA activity. In the future, the generalized newborn screening for lysosomal storage disorders should become a part of the standard routine. Therefore, it will be advisable to adopt on a greater scale more automated procedures, such as mass spectrometry. In this case study, performed on urine samples taken from patients with infantile, juvenile and adult form of Pompe disease before and during/after ERT, we show the application of the highly specific and sensitive MALDI-TOF MS method for Glc4 detection, and we propose using NMR as a precise method for its quantification. The advantages and disadvantages of both methods are also discussed.
Experimental
Materials and reagents
Standard glucose tetrasaccharide (Glc4 standard), ammonium dihydrogen phosphate (NH4H2PO4), 3-aminoquinoline (3-AQ), acetonitrile (ACN), and water in LC–MS qualities were purchased from Sigma Aldrich (St. Louis, MO, USA). Sodium tetradeuterium trimethylsilylpropionate (TSPd4) and deuterated water (D2O) were purchased from Eurisotop (Saint-Aubin Cedex, FR). α-Cyano-4-hydroxycinnamic acid (CHCA) was obtained from Bruker Daltonics (Billerica, MA, USA) and trifluoroacetic acid (TFA) was purchased from Fluka (St. Gallen, Switzerland).
Samples
Samples of four anonymized patients treated by ERT, previously diagnosed with Pompe diagnosis by GAA activity assay in peripheral leucocytes, and four healthy anonymous donors, as negative controls, were analyzed. Samples were obtained from National Institute of Children’s Diseases, in accordance with standard operating procedures. Written informed consent was given by every subject and the experimental study protocol was reviewed and confirmed by the hospital ethics committee.
Patient 1—male, infantile IOPD form, age < 1 year at the time of the last urine sample collection. At the age of 8 months, the first symptoms of respiratory track infection and congestive heart failure appeared. After ERT onset, his health status was stabilized. Muscle strength and the psycho-motor development of the child became adequate for his age. Developmental milestones, previously lagging, became normal. Cardiological findings showed good function of both atria in the heart. Neurological findings, including electromyography (EMG), were normal. Urine was collected prior to ERT onset, after 2 weeks and after 1 month of treatment.
Patient 2—female, juvenile LOPD form, 7 years old at the time of the last urine sample collection. Before ERT, the patient was tired and unable to play with her coevals. After beginning ERT, her fatigue no longer troubled her; she started to attend school, and had enough energy to participate in all activities with the other children. Urine was collected prior to starting ERT, after 5 and after 8 months of the ERT treatment.
Patient 3—male, juvenile LOPD form, samples taken at the age of 4 and 7 years. At the age of 4 years, the child did not show any clinical symptoms in spite of elevated Glc4 levels and GAA deficiency; therefore, the therapy was not initiated. A second sample of urine was taken at the age of 7 years, when clinical symptoms appeared. Laboratory tests confirmed increased values of creatine kinase (CK) and lactate dehydrogenase (LD). Clinical signs of a worsening of muscle function were manifested by an atypical gait and the necessity of using his hands when standing up (Gower’s sign). In comparison with Patient 2, his motor ability was worse. His cardiological status was without any pathology.
Patient 4—male, adult LOPD form, age > 25 years at the time of the last urinary sample collection. Before beginning ERT, the patient could walk with a walking frame; afterwards, he was able to walk alone and could manage all everyday personal care by himself. After declining further ERT, he became disabled once more and had to use a wheel chair. Urine samples were collected after 1 year of ERT and 3 years after ERT rejection.
The urine samples of all 4 patients were frozen immediately after their collection and stored at – 20 °C until the analysis was performed. The samples of urine from both the patients and the anonymized healthy volunteers were analyzed under the same conditions.
Methods
MALDI-TOF MS
Glc4 standard solution was prepared in a concentration of 1 mg.mL−1 using ACN/water (1:1, v/v) with the addition of TFA to the final concentration of 0.1% (v/v). The method based on direct on-target derivatization (Kaneshiro et al. 2011) was used. A saturated solution of CHCA was prepared by dissolving 10 mg of CHCA in 540 μL of ACN/water (1:1, v/v) and 60 μL of 100 mM NH4H2PO4 aqueous solution. A 3-AQ/CHCA mixture was prepared by dissolving 20 mg of 3-AQ in 150 μL of saturated CHCA solution and finally diluted tenfold using ACN/water (1:1, v/v) to obtain a working 3-AQ/CHCA matrix solution. 0.5 μL of tenfold diluted urine or standard Glc4 solution and 0.5 μL of a 3-AQ/CHCA working solution were premixed on a ground steel MALDI target plate (Bruker Daltonics, MA, USA). Afterwards, the plate was incubated for 60 min at 60 °C. On-target derivatized samples were analyzed by UltrafleXtreme TOF/TOF mass spectrometer (Bruker Daltonics, MA, USA) in the reflectron positive ion mode for MS and the LIFT ion mode for MS/MS analysis. External calibration was performed by Peptide calibration standard II (Bruker Daltonics, MA, USA). Raw spectra were processed using the FlexAnalysis software (Bruker Daltonics, USA) with baseline subtraction and spectral smoothing.
1H NMR spectroscopy
100 µL of urine was freeze-dried and re-dissolved in 250 µL of 0.4 mM TSPd4 in D2O. D2O was used for magnetic field locking and shimming. Trimethylsilyl propionic acid (TSPd4) was used for chemical shift set up (δ = 0 ppm), as well as the internal standard for Glc4 oligosaccharide quantification. NMR spectra were measured at 600 MHz on a Varian VNMRS NMR spectrometer equipped with triple resonance HCN, a 13C enhanced salt tolerant cold probe head, at 25 °C in 3 mm sample tubes. The residual water signal was suppressed using PRESAT sequence with 30° 1H pulse, 5 s acquisition time, and 2 s relaxation delay. Anomeric proton signals in the 1H NMR spectra were assigned according to the literature (Pakanová et al. 2016).
From 1H NMR spectrum, the concentration of metabolite is calculated relatively to the internal standard (added at a known concentration) as follows:
where [m] is the concentration of metabolite m to be quantified; A0 is the area of m metabolite signal in the spectrum; Aref is the area of TSPd4 resonance; and 9 and b are the numbers of protons in TSPd4 and in the metabolite m signals, respectively.
Results and discussion
3-AQ derivatization and MALDI-TOF MS analysis
Using MALDI-TOF MS for the fast screening of oligosaccharidoses without pre-treatment was described by Bonneso et al. (2014). They performed urinary screening for various oligosaccharidoses in a single rapid step using 3-AQ as a common MALDI matrix. However, in some cases, the derivatization of the sample is preferred. In our study, we have utilized an ability of 3-AQ to bind covalently to free, reducing ends of sugars during on-plate incubation at 60 °C, to increase sensitivity and selectivity. Increasing the sensitivity is important, especially in cases, where the Glc4 levels may be low, in mild forms of Pompe disease, or after the long-term administration of enzyme therapy. This targeted derivatization of saccharide molecules was applied to the urine samples of the Pompe patients (n = 4), healthy subjects (n = 4), and the Glc4 standard. After the derivatization, the positive ion mode in the MALDI-TOF measurement showed adduct molecules, 3-AQ derivatized saccharides, with increased mass by 126 Da (Kaneshiro et al. 2011). Thus, in the MALDI-TOF MS spectra of the Pompe patients´ urine, instead of the signal of the sodiated Glc4 (m/z 689.2), a 3-AQ derivatized Glc4 adduct (m/z 815.2) appeared (Fig. 1). These 126 Da adduct molecules are formed by a nonreductive amination of the saccharides by 3-AQ at their reducing ends. The formation of such adduct molecules was reported by Rohmer et al. 2010. Selective 3-AQ derivatization of urinary oligosaccharides leads to the increased specificity of the method.

Reflectron positive MALDI-TOF MS spectra of Glc4 standard before (“Free U-Glc4 standard”) and after (“3-AQ derivatized U-Glc4 standard”) the derivatization with highlighted 126 Da mass increment. Signals with m/z 689.199 and 815.182 represent the sodiated adduct of Glc4 tetrasaccharide. 3-AQ—3-aminoquinoline
Representative MS spectra of Glc4 standard and the urine of the Pompe patients and healthy individuals are shown in Fig. 2. To confirm the presence of the Glc4 structure, MS/MS (LIFT) analysis of the sodiated parent ion at m/z 815.2 was performed and the fragmentation pattern was compared to that of the Glc4 standard (Fig. 3). Predominantly, “bi” (containing the non-reducing end) and “yj” (containing the 3-AQ labeled reducing end) fragment ions were formed, creating a typical pattern of MALDI-TOF/TOF fragmentation of saccharides. The subscript i indicates the number of glycosidic bonds cleaved, counted from the non-reducing end, and j refers to the number of inter glycosidic bonds counted from the reducing end (Domon and Costello 1988; Bauer. 2012). In addition to the 3-AQ derivatized parent ion (m/z 815.2) of the Glc4, several lower intensity signals, representing oligosaccharides with longer chains (up to Glc11), were also detected (i.e., Glc7 shown in the spectra with m/z 1301.3). The presence of such oligosaccharides in the urine of Pompe patients was also confirmed by Rozaklis et al. 2002.

Reflectron positive MALDI-TOF MS spectra of 3-AQ-treated urinary samples from Pompe patients without ERT (n = 4), Glc4 standard as a positive control, and urine samples from healthy individuals (n = 4) as negative controls. The depicted signal at m/z 815.2 represents the sodiated form of 3-AQ derivatized Glc4, which was present only in the urines of Pompe patients. 3-AQ 3-aminoquinoline, ERT enzyme replacement therapy, Patient 1—Infantile Onset Pompe Disease (IOPD) before ERT; Patient 2 and 3—Juvenile Late Onset Pompe Disease (LOPD) before ERT; Patient 4—Adult LOPD after ERT; Healthy 1–4—urinary samples from healthy individuals; ERT enzyme replacement therapy

Representative MALDI TOF/TOF (LIFT) spectra of 3-AQ derivatized parent ion with m/z 815.2 issued from Glc4 tetrasaccharide standard and urine of Pompe patient 1 [Infantile Onset Pompe Disease (IOPD)] before the ERT onset. Typical “b” (containing the non-reducing end) and “y” (containing the 3-AQ labeled reducing end) fragmentation ions are formed predominantly. Hex hexose unit (i.e., glucose); 3-AQ 3-aminoquinoline
In general, the determination of quantity by MS analysis is challenging owing to numerous factors, which include: (i) the need for internal standards or structural analogues (ideally isotopically labeled); (ii) the fact that such standards may be unavailable or prohibitively costly; (iii) the need to maintain the standards’ concentrations close to those of the target metabolites; and (iv) the alternative use of time-consuming calibration curves for each target metabolite. Mass spectrometry lacks the capability of enabling a simple absolute quantification using a single internal standard, due to a number of factors, including ionization efficiencies, ion suppression, and matrix effects (Gowda et al. 2018). In the case of complex matrices, such as urine, and due to the wide concentration ranges of Glc4 in individual patients, we decided to use NMR for more precise quantification.
1H NMR analysis and Glc4 quantification
Besides the mass spectrometry, NMR is another important analytical technique used for the characterisation of complex biological matrices in metabolomic studies. NMR spectroscopy is not destructive; it provides quantitative spectra reflecting the concentration of many metabolites simultaneously. Usually, it does not require a sample treatment before the measurement. In complex NMR spectra, a signal position depends on its chemical environment and also often on pH values, and the signal intensity directly reflects the quantity of metabolite in the body fluid. In the 1H NMR spectrum, Glc4 tetrasaccharide (αGlc(1 → 6)αGlc(1 → 4)αGlc(1 → 4)α,βGlc; 6-α-D-glucopyranosyl-maltotriose) identification is based on the characteristic chemical shifts of anomeric H1 signals of each glucose unit (Kumlien et al. 1989). The anomeric H1 signal coming from the terminal Glc residue (δ 4.96 ppm), which is linked to the O6 of the neighbouring internal 1,4-linked αGlc, is the most specific for Glc4 quantification, as can be seen in Fig. 4 (Pakanová et al. 2016). The signals of two internal 1,4-linked αGlc residues in Glc4 are partially overlapped at δ 5.410, 5.401. Glc4 quantification based on these signals might be possible, but it will suffer from inaccuracy caused by an overlap with the H1 issuing from the maltodextrins (M4), which should account for ~ 5% of the total Hex4 (Young et al. 2012). Figure 4 shows the α anomeric region of proton spectra measured from urine of five Pompe patients, Glc4 standard and two healthy subjects. In the most of the spectra, the signal due to α-1,6-linked Glc (δ 4.969–4.967) is well resolved. A low variation of chemical shifts in the spectra is caused by the differences in pH of urine solutions. Only in two spectra of urine, Patient 1 (2 weeks on ERT) and Patient 4 (3 years after the refusal of ERT), these signals are overlapped. In the former case, only one half of the H1 doublet signal was visible (Fig. 4, marked as A) and it has served for the precise Glc4 quantification (its integral value was multiplied by two in the Glc4 quantity calculations). In the second case, the overlap of α-1,6-linked Glc was complete. However, the signals due to the α-1,4-linked glucose units could be identified (marked as B) and it was used for the Glc4 level determination. The possibility of presence of other signals in their proximity, which were observed in all other spectra (Fig. 4, dotted grey lines), increases the degree of inaccuracy of determined Glc4 concentration values. Thus, the possible presence of maltodextrin (< 10–15%) should be taking into account.

Anomeric region of the 1H NMR spectra of Pompe patients’ urines and Glc4 standard. Examined urine samples were collected before ERT onset and after various time periods of ERT administration (w week, m month). ERT enzyme replacement therapy; Patient 1—Infantile Onset Pompe Disease (IOPD); Patients 2 and 3—Juvenile Late Onset Pompe Disease (LOPD); Patient 4—Adult form of LOPD. 1,4αGlc—1,4-linked glucose; 1,6αGlc—terminal Glc of the tetrasaccharide linked to the O6 of the neighbouring α-Glc. According to Table 1: A—one half of the doublet was used for quantification (Patient 1, ERT 2w), B—used for quantification (Patient 4, post ERT)
1H NMR spectroscopy is frequently used in metabolomics studies. It is a highly quantitative method which enables the detection of protons of all metabolites present in a biofluid at the moment of its collection. Signal intensities depend on the number of protons resonating at a given frequency and the concentration of metabolites. Their absolute quantity may be obtained by different modes. In the frequently used indirect mode, the signal intensity of the biomarker is compared with the signal of an internal standard, which resonates outside of the spectral regions of interest. Knowledge about the amount of accumulated oligosaccharides could be useful for follow-up of the treatment and would require an internal standard. For this purpose, quantities should be compared to the creatinine (CRN) level. Creatinine is a variable parameter which can be used to measure the urine concentration and which is often used as a correcting factor in many analytical methods involving urine (Bonesso et al. 2014). Its signal is well resolved in the 1H NMR spectra of urines and thus quantities of both creatinine and biomarkers can be found in one spectrum.
Analysis of the 1H NMR spectra of the Patient 1 urine (IOPD) before the ERT onset contained 63.7 mM/M creatinine (CRN) of Glc4. Two weeks after ERT onset, the Glc4 value had decreased to 26.2 mM/M CRN, and after 1 month of ERT, the Glc4 level was 12.6 mM/M CRN. This value was lower than the maximum value found for age-matched control patients (Sluiter et al. 2012; Young et al. 2012; Lobato et al. 2013).
Patient 2, with juvenile LOPD, matched the age group of 4–10 years. ERT treatment was found to be very efficient as the level of Glc4 decreased from 38.5 to 26.4 and 7.8 (all in mM/M CRN) after 5 and 8 months, respectively. Comparison with the Glc4 levels of age-matched negative controls showed that the Glc4 level after 8 months of ERT was slightly lower than the median values found by Sluiter et al. 2012, and slightly higher than the maximum value found by An et al. 2000.
Patient 3, with juvenile LOPD, also matched the age group of 4–10 years. In his first urine sample, at age 4 years, the Glc4 level was 45.95 mM/L CRN. At that time, he did not show any clinical signs of Pompe disease in spite of low GAA activity and an elevated Glc4 level, and thus, he did not receive ERT. At present, at the age of 7 years, clinical signs have appeared. 1H NMR showed that the Glc4 concentration had increased to 66.76 mM/M CRN, confirming that ERT should start immediately.
Patient 4, with the adult form of LOPD, had been receiving ERT for 1 year when his urine was analyzed for its Glc4 level. The Glc4 level of 6.6 mM/M CRN was very close to the median value of Pompe patients found by Sluiter et al. 2012, but was still high when compared with the values found for control individuals. This patient decided to stop the therapy and the analysis performed 3 years after cessation of the therapy showed, besides a worsening of clinical symptoms, that the Glc4 level had increased to 16.7 mM/M CRN (Table 1).
Widely used diagnostic methods such as HPLC–MS usually require pre-treatment of urinary samples by derivatization followed by a clean-up procedure. The use of a powerful mass analyser with MS/MS (triple quadrupole, TOF/TOF) or MSn (ion trap) capabilities, in conjunction with reverse-phase, normal phase, porous graphited carbon, size exclusion, ion exchange, liquid chromatographic, or capillary electrophoretic separation methods has increased the use of MS in characterizing oligosaccharide structures. The main function of these coupled techniques is to reduce complexity by separating isobaric structures that are not resolved by MS. During the derivatization reaction, salts, solvents, and derivatization reagents are present in excessive amounts, and a clean-up procedure must be implemented to remove the excess to facilitate oligosaccharide detection (Casado et al. 2017). Another strategy is to develop a simple analytical method to identify the metabolite without any pre-treatment. The limitations of such an approach appear when the intensity of the metabolite signals is close to the signal-to-noise threshold. During long-term ERT administration, or in mild cases of Pompe disease, Glc4 levels can decrease to almost undetectable values. Thus, for the identification of Glc4, derivatization may be required. Nevertheless, in our study, we have confirmed that on-target 3-AQ derivatization has its major advantage in the reduction of the potential sample or its components loss and is thus useful for fast and robust screening for increased Glc4 levels in the urine samples of Pompe patients. When using this method, no purification or separation steps are required. This quick and easy operational protocol decreases the time and cost of analysis. Two further advantages of on-plate derivatization are the possibility of automatization (as the MALDI target plates consist of 384 spots) and the fact that only 0.05 microliters of urine are necessary per one analysis.
We have also compared our method with that published by Bonesso et al. 2014, in which 3-AQ was used as the matrix. For the 3-AQ derivatization of urinary oligosaccharides by our method, sample amounts 3 times lower were required, plus significantly lower laser intensities were used for the acquisition of spectra of good quality (data not shown). It is well known that in the case of MALDI measurements, the laser settings should be kept at the lowest possible intensity to preserve ions and to avoid a typical in-source fragmentation. Thus, the direct on-plate derivatization led to an overall improvement of this diagnostic method. However, the main disadvantage of MALDI-TOF MS in diagnostics of Pompe disease is its non-quantitative nature. The addition of isotopically labeled internal standards could solve this problem, but it would dramatically increase costs per analysis. Furthermore, as the Glc4 concentrations vary in individual patients, an effort to maintain the standards’ concentrations close to those of the target metabolites could be challenging. Moreover, ionization efficiencies can differ significantly and, together with the fact that other compounds present in the biofluid may compete or interfere with ionization (Bauer. 2012), they make the quantification of oligosaccharides by mass spectrometry difficult.
Contrary to MALDI, NMR spectroscopy is able not only to detect but also to quantify Glc4 levels in Pompe patients’ urine. Its advantages are: the single measurement is very fast (spectrum measured within 15–30 min); the method is higly reproducible and not destructive; and the creatinine quantity can be calculated from the same spectrum. Its disadvantages: less sensitivity resulting in bigger urine volume for analysis than for MALDI; possible overlap of the H1 signal of 1,6-linked terminal α-Glc used for the quantification with signals of other metabolites present in urine. In the latter case, quantification might still be possible if the signals of 1,4-linked α-Glc units are well separated. Such quantification suffers from small error caused by the presence of α-dextrins (5% of the total Hex4, Young et al. 2012). The degree of inaccuracy increases if other signals are observed in their close proximity.
In spite of the fact that the efficacy of ERT is variable and unpredictable, especially in patients with LOPD (Dasouki et al. 2014), decreased Glc4 levels in two patients under ERT involved in our study have confirmed its efficiency. In the case of Patient 3 with delayed onset of clinical signs, the determination of Glc4 levels by NMR confirmed his suitability to begin immediate enzymatic therapy. In the case of Patient 4 after therapy rejection, besides the worsening of clinical signs, NMR clearly showed a significant increase in Glc4 levels. However, the sensitivity of NMR, at the magnetic field frequency and with the cryo probe used for the measurements, was not sufficient for Glc4 detection in urine collected from healthy volunteers.
Nowadays, there are alternative therapies under development for Pompe disease (Schoser et al. 2017). These include second generation ERTs that aim to improve targeting and extracellular uptake of the recombinant protein, either alone or in combination with pharmacological chaperones; oligonucleotide therapeutics in the form of mRNA-delivered GAA or exon-including antisense RNA (Safdar et al. 2016; Bergsma et al. 2016). Furthermore, gene therapies are another route of treatment, seeking to continuously and endogenously produce GAA in target tissues and possibly minimize anti-GAA immunogenicity (Xu et al. 2005; So et al. 2010; van Til et al. 2010). Future determination of urinary Glc4 levels in Pompe patients undergoing such modern therapeutic approaches could help us understand its prospective efficiency.
Conclusions
This study presents an application of the mass spectrometry and NMR spectroscopy to the urine analysis of Pompe patients, focused on Glc4 detection and quantification. Both methods can shorten the duration of the diagnostic process and are highly accurate. The results obtained show that, due to their robustness, selective on-target 3-AQ derivatization of oligosaccharides in urine with MALDI-TOF/TOF detection and 1H NMR spectroscopy is suitable for IOPD and juvenile LOPD Pompe disease screening in the young population. Unlike MALDI-TOF MS, the 1H NMR spectroscopy not only confirms the presence of Glc4 but also quantifies the Glc4, and so can be effectively utilized in the monitoring of ERT efficiency. Both methods are complementary (mass spectrometry is highly sensitive and NMR spectroscopy is highly quantitative) and they significantly contribute to Pompe disease diagnostics by the fast estimation of Glc4 presence, which may help prevent a high degree of damage to Pompe patient physiology. The results of our study suggest that on-target 3-AQ derivatization of urinary oligosaccharides with MALDI-TOF MS detection is the most appropriate method for quick screening and NMR is similarly suitable for further extended evaluation of the changes of Glc4 levels during long-term enzymatic therapy.
References
Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT (2001) Recombinant human acid a-glucosidase enzyme therapy for infants with glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 3:132–138
An Y, Young SP, Hillman SL, van Hove JLK, Chen YT, Millington DS (2000) Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem 287(1):136–143. https://doi.org/10.1006/abio.2000.4838
An Y, Young SP, Kishnani PS, Millington DS, Amalfitato A, Corzo D, Chen YT (2005) Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disese. Mol Genet Metabol 85(4):247–254. https://doi.org/10.1016/j.ymgme.2005.03.010
Bauer S (2012) Mass spectrometry for characterizing plant cell wall polysaccharides. Front Plant Sci 3:45. https://doi.org/10.3389/fpls.2012.00045
Bergsma AJ, In’t Groen SL, Verheijen FW, van der Ploeg AT, Pijnappel WP (2016) From cryptic toward canonical pre-mRNA splicing in Pompe disease: a pipeline for the development of antisense oligonucleotides. Mol Ther Nucleic Acids 5(9):e361. https://doi.org/10.1038/mtna.2016.75
Bonesso L, Piraud M, Caruba C, van Obberghen E, Mengual R, Hinault Ch (2014) Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry. Orph J Rare Dis 9:19. https://doi.org/10.1186/1750-1172-9-19
Burton BK (2012) Newborn screening for Pompe disease: an update. Am J Med Genet C Semin Med Genet 160C(1):8–12. https://doi.org/10.1002/ajmg.c.31315
Casado M, Altimira L, Montero R, Castejón E, Nascimento A, Pérez-Duenas B, Ormazabal A, Artuch R (2014) A capillary electrophoresis procedure for the screening of oligosaccharidoses and related diseases. Anal Bioanal Chem 406(18):4337–4343. https://doi.org/10.1007/s00216-014-7832-6
Casado M, Ferrer-López I, Ruiz-Sala P, Pérez-Cerdá C, Artuch R (2017) Urine oligosaccharide tests fot the diagnosis of oligosaccharidoses. Rev Anal Chem. https://doi.org/10.1515/revac-2016-0019
Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL (2009) Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 124(6):e1116–e1125. https://doi.org/10.1542/peds.2008-3667
Chien YH, Lee NCh, Chen ChA, Tsai FJ, Tsai WH, Shieh JY, Huang HJ, Hsu WCh, Tsai TH, Hwu WL (2015a) Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr 166(4):985–991.e1–2. https://doi.org/10.1016/j.jpeds.2014.10.068
Chien YH, Goldstein JL, Hwu WL, Smith PB, Ni-Chung L, Chiang SCh, Tolun AA, Zhang H, Vaisnins AE, Millington DS, Kishnani PS, Young SP (2015b) Baseline urinary glucose tetrasaccharide concentrations in patients with infantile- and late-onset Pompe disease identified by newborn screening. JIMD Reports 19:67–73. https://doi.org/10.1007/8904_2014_366
Dasouki M, Jawdat O, Amdhoun O, Pasnoor M, McVey AL, Auzinadah A, Herbelin L, Barohn J, Dimachkie MM (2014) Pompe disease: literature review and case series. Neurol Clin 32(3):751–776. https://doi.org/10.1016/j.ncl.2014.04.010
de Almeida Thomazinho P, Scalco FB, de Oliveira MLC, Horovitz DDG, Llerena JC Jr (2017) Motor development as a potential marker to monitor infantile Pompe disease on enzyme replacement therapy. Open J Clin Diagnostics 7(1):8–19. https://doi.org/10.4236/ojcd.2017.71002
Domon B, Costello CE (1988) A systematic nomenclature for carbohydrate fragmentation in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate J 5(4):397–409. https://doi.org/10.1007/BF01049915
Gowda GAN, Djukovic D, fan Bettcher L, Gu H, Raftery D (2018) NMR-guided mass spectrometry for absolute quantitation of human blood metabolites. Anal Chem 90:2001–2009. https://doi.org/10.1021/acs.analchem.7b04089
Hallgren P, Hansson G, Henriksson KG, Häger A, Lundblad A, Svensson S (1974) Increased excretion of a glucose-containing tetrasaccharide in the urine of a patient with glycogen storage disease type II (Pompe’s disease). Eur J Clin Investig 4(6):429–433
Huang CK, Liao HC, Hsieh YP, Chen YC, Yang FC, Niu DM (2015) Glucose tetrasaccharide (Glc4) level in urine sample as a biomarker for Pompe patients. Ann Transl Med 3(S2):AB067. https://doi.org/10.3978/j.issn.2305-5839.2015.ab067
Kaneshiro K, Fukuyama Y, Iwamoto S, Sekiya S, Tanaka K (2011) Highly sensitive MALDI analyses of glycans by a new aminoquinoline-labeling method using 3-aminoquinoline/α-cyano-4-hydroxycinnamic acid liquid matrix. Anal Chem 83(10):3663–3667. https://doi.org/10.1021/ac103203v
Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg AT, Clancy JP, Parini R, Morin G, Beck M, de la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE (2007) Recombinant human acid alpha-glucosidase: major clinical benefits in infantile onset Pompe disease. Neurology 68(2):99–109. https://doi.org/10.1212/01.wnl.0000251268.41188.04
Kishnani PS, Amartino HM, Lindberg Ch, Miller TM, Wilson A, Kreutzer J (2014) Methods of diagnosing of patients with Pompe disease: data from the Pompe registry. Mol Gen and Metabolism 113(1–2):84–91. https://doi.org/10.1016/j.ymgme.2014.07.014
Kumlien J, Grönberg G, Nilsson B, Mänsson O, Zopf D, Lundblad A (1989) Structural and immunochemical analysis of three α-limit dextrin oligosaccharides. Arch Biochem Biophys 269(2):678–689
Kuperus E, Kruijshaar ME, Wens SCA, de Vries JM, Favejee MM, van der Meijden Rizopoulos, Brusse E, van Doorn PA, van der Ploeg AT, van der Beek N (2017) Long-term benefit of enzyme replacement therapy in Pompe disease. A 5-year prospective study. Neurology 89(23):2365–2373. https://doi.org/10.1212/wnl.0000000000004711
Lim JA, Li L, Raben N (2014) Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci 6:177. https://doi.org/10.3389/fnagi.2014.00177
Lobato JB, Parejo PD, Diez PT, Jimenez LM (2013) Glucosa tetrasacárido como biomarcador diagnóstico de la enfermedad de Pompe: estudio en 35 pacientes. Med Clin Barc 141(3):106–110. https://doi.org/10.1016/j.medcli.2013.02.035
Manwaring V, Prunty H, Bainbridge K, Burke D, Finnegan N, Franses R, Lam A, Vellodi A, Heales S (2012) Urine analysis of glucose tetrasaccharide by HPLC; a useful marker for the investigation of patients with Pompe and other glycogen storage diseases. J Inh Metab Dis 35:311–316. https://doi.org/10.1007/s10545-011-9360-2
Pakanová Z, Matulová M, Behúlová D, Šalingová A, Hlavatá A, Pätoprstý V, Mucha J (2016) Molecular diagnosis of Pompe disease using MALDI TOF/TOF and 1H NMR. Chem Pap 70(3):265–271. https://doi.org/10.1515/chempap-2015-0218
Pérez-López J, Selva-O’Callaghan A, Grau-Junyent JM, Gallego-Galindo L, Coll MJ, García-Morillod S, Torralba-Cabeza MA, Vilardell-Tarrés M (2015) Delayed diagnosis of late-onset Pompe disease in patients with myopathies of unknown origin and/or hyperCKemia. Mol Genet Metab 114(4):580–583. https://doi.org/10.1016/j.ymgme.2015.02.004
Rohmer M, Meyer B, Mank M, Stahl B, Bahr U, Karas M (2010) 3-Aminoquinoline acting as matrix and derivatizing agent for MALDI MS analysis of oligosaccharides. Anal Chem 82(9):3719–3726. https://doi.org/10.1021/ac1001096
Rozaklis T, Ramsay SL, Whitfield PD, Ranieri E, Hopwood JJ, Meikle PJ (2002) Determination of oligosaccharides in Pompe disease by electrospray ionization tandem mass spectrometry. Clin Chem 48(1):131–139
Safdar A, Nilsson M, Akhtar M, Tarnopolsky M (2016) Exosome-mRNA (EXERNA) therapy for Pompe disease. Mol Genet Metab 117:S100. https://doi.org/10.1016/j.ymgme.2015.12.422
Schielen PCJI, Kemper EA, Gelb MH (2017) Newborn screening for lysosomal storage diseases: a concise review of the literature on screening methods, therapeutic possibilities and regional programs. Int J Neonat Screen 3(2):6. https://doi.org/10.3390/ijns3020006
Schoser B, Bilder DA, Dimmock D, Gupta D, James ES, Prasad S (2017) The humanistic burden of Pompe disease: are there still unmet needs? A systematic review. BMC Neurol 17:202. https://doi.org/10.1186/s12883-017-0983-2
Sluiter W, van den Bosch JC, Goudriaan DA, van Gelder CM, de Vries JM, Huijmans JGM, Reuser AJJ, van der Ploeg AT, Ruijter GJG (2012) Rapid ultraperformance liquid chromatography–tandem mass spectrometry assay for a characteristic glycogen-derived tetrasaccharide in Pompe disease and other glycogen storage diseases. Clin Chem 58(7):1139–1147. https://doi.org/10.1373/clinchem.2011.178319
So K, Iizuka S, Kobayashi H, Kimura T, Fukuda T, Shen J, Shimada Y, Ida H, Eto Y, Ohashi T (2010) Neonatal gene transfer using lentiviral vector for murine Pompe disease: long-term expression and glycogen reduction. Gene Ther 17(4):521–530. https://doi.org/10.1038/gt.2009.160
Strothotte S, Strigl-Pill N, Grunert B, Kornblum C, Eger K, Wessig C, Deschauer M, Breunig F, Glocker FX, Vielhaber S, Brejova A, Hilz M, Reiners K, Müller-Felber W, Mengel E, Spranger M, Schoser B (2010) Enzyme replacement theraphy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12 month results of an observational clinical trial. J Neurol 257(1):91–97. https://doi.org/10.1007/s00415-009-5275-3
Szájli E, Fehér T, Medzihradszky KF (2008) Investigating the quantitative nature of MALDI-TOF MS. Mol Cel Proteomics 7(12):2410–2418. https://doi.org/10.1074/mcp.M800108-MCP200
van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, de Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, van Hirtum H, van Diggelen OP, Boer M, Kroos MA, van Doorn PA, van der Voort E, Sibbles B, van Corven EJ, Brakenhoff JP, van Hove J, Smeitink JA, de Jong G, Reuser AJ, van der Ploeg AT (2004) Long-term intravenous treatment of Pompe disease with recombinant human alfa-glucosidase from milk. Pediatrics 113(5):e448–e457
van der Ploeg AT, Reuser AJ (2008) Pompe’s disease. Lancet 372(9646):1342–1353. https://doi.org/10.1016/S0140-6736(08)61555-X
van Til NP, Stok M, Aerts Kaya FS, de Waard MC, Farahbakhshian E, Visser TP, Kroos MA, Jacobs EH, Willart MA, van der Wegen P, Scholte BJ, Lambrecht BN, Duncker DJ, van der Ploeg AT, Reuser AJ, Verstegen MM, Wagemaker G (2010) Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 115(26):5329–5337. https://doi.org/10.1182/blood-2009-11-252874
Xia B, Asif G, Arthur L, Pervaiz MA, Li X, Liu R, Cummings RD, He M (2013) Oligosaccharide analysis in urine by MALDI-TOF mass spectrometry for the diagnosing of lysosomal storage diseases. Clin Chem 59(9):1357–1368. https://doi.org/10.1373/clinchem.2012.201053
Xu F, Ding E, Migone F, Serra D, Schneider A, Chen YT, Amalfitano A (2005) Glycogen storage in multiple muscles of old GSD-II mice can be rapidly cleared after a single intravenous injection with a modified adenoviral vector expressing hGAA. J Gene Med 7(2):171–178. https://doi.org/10.1002/jgm.660
Young SP, Zhang H, Corzo D, Thurbewrg BL, Bali D, Kishnani PS, Millington DS (2009) Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet Med 11(7):536–541. https://doi.org/10.1097/GIM.0b013e3181a87867
Young SP, Piraud M, Goldstein JL, Zhang H, Rehder C, Laforet P, Kishnani PS, Millington DS, Bashir MR, Bali DS (2012) Assessing disease severity in Pompe disease: the roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am J Med Genet Part C Semin Med Gen 160C(1):50–58. https://doi.org/10.1002/ajmg.c.31320
Acknowledgements
Financial support from VEGA 2/0130/18, VEGA 2/0188/14 and APVV-15-0410 grant are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pakanová, Z., Matulová, M., Uhliariková, I. et al. Case study: monitoring of Glc4 tetrasaccharide in the urine of Pompe patients, use of MALDI-TOF MS, and 1H NMR. Chem. Pap. 73, 701–711 (2019). https://doi.org/10.1007/s11696-018-0623-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-018-0623-3