
Abstract
In the present study, Ni-P-ZnO nanocomposite coating was developed on the surface of mild steel substrate by electroless technique. The second phase ZnO nanoparticles were synthesized by autocombustion method, to incorporate into the Ni-P matrix. 10 g/L of ZnO nanoparticles were added to the Ni-P alkaline bath for co-deposition, and the electroless bath was reduced by sodium hypophosphite. The heat treatment of the as-prepared Ni-P/Ni-P-ZnO coatings was carried out at 400 °C in argon (99.9%) atmosphere for 1 h. The as-prepared and heat-treated Ni-P/Ni-P-ZnO coatings and ZnO nanoparticles were analyzed for surface morphology, elemental composition, phase analysis and particle size distribution using field emission scanning electron microscopy (FESEM), energy-dispersive analysis of x-rays (EDAX), transmission electron microscopy (TEM), powder x-ray diffraction analysis. To determine the calcinations temperature of ZnO powder, differential scanning calorimetry was also carried out. TEM analysis of the synthesized ZnO nanoparticles was also carried out, and from the micrographs a spherical shape of ~40 nm size range is observed for the ZnO nanoparticles. The corrosion properties of the coatings were carried out in a 3.5 wt.% NaCl solution by electrochemical polarization test. The dispersion of ZnO nanoparticles into the coating is determined by FESEM. Atomic force microscopy was used to investigate the change in the surface topography of the coatings before and after exposure in the test environment. A uniform distribution of ZnO nanoparticles into the Ni-P matrix is confirmed by the FESEM-EDAX results. Electrochemical test results suggest that Ni-P-ZnO shows better corrosion resistance as compared to plain Ni-P coating.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Electroless coating has gained a wide spread interest by researchers owing to its improved properties with less complicated setup. Unlike electroplating, it is a controlled chemical reduction process in which several chemical reactions take place simultaneously, in aqueous medium without the use of electric current. In electroless coating technique, there is uniformity in the composition and thickness of the coating as well as the intricate part of the substrate has equal opportunity to be coated (Ref 1-3). By electroless coating technique pure metallic Ni, binary alloy (Ni-P (Ref 1-4) and Ni-B (Ref 1, 5, 6), Co-P (Ref 2, 3) and Co-B (Ref 2, 3), etc.) and ternary alloy (Ni-P-B (Ref 3, 7), Ni-W-P (Ref 8), Ni-Co-P (Ref 9, 10), etc.) were coated successfully and studied for their tribological properties. For the last two decades, the emphasis has been shifted toward co-deposition of second phase particles into the Ni-P matrix to tailor their properties for different industrial applications. Extensive research was carried out to control the parameters for co-deposition of second phase nanoparticles in the electroless Ni-P matrix and led this old electroless coating technology to a newer step, i.e., electroless nanocomposite coatings. Several particles were incorporated in the nickel matrix, and the choice of the co-deposition of second phase (X) elements depends upon the application. For example, when high wear resistance properties are the main requirement then hard particles, viz., SiC, WC, Al2O3 and Si3N4, etc., are preferred for co-deposition (Ref 11-15). Out of Ni-P-X (where X = hard particles), Ni-P-SiC coatings show high hardness and wear resistance in order to replace existing “hard chromium” plating in the aerospace industry (Ref 13-15). Among others, this combination (Ni-P-SiC coatings) has proved a cost-effective and best-performing combination applied in industry.
Dong et al. (Ref 16) reported that incorporation of SiO2 nanoparticles in electroless Ni-P coating significantly improves the microhardness (on heat treatment at ~400 °C) and wear resistance of the coatings. The corrosion resistance property of electroless Ni-P-SiO2 coatings against salty solutions was significantly improved, where SiO2 is (2 wt.%; size ~20 nm) (Ref 16, 17). To develop Ni-P-TiO2 composite coating, a novel technique, i.e., addition of transparent TiO2 sol into the conventional electroless plating solution was reported (Ref 18). Microhardness test results show a significant improvement from ~710 HV0.2 to 1025 HV0.2 for conventional Ni-P-TiO2 coating (Ref 18).
Furthermore, soft particles such as PTFE, hexagonal boron nitride (BN (h)), molybdenum disulfide (MoS2), inorganic fullerene-tungsten disulfide (WS2), carbon nanotubes and graphite (C) provide good lubrication and are capable of providing a low average Index number (Iav) when incorporated in the electroless Ni-P matrix (Ref 19-24). These soft/lubricating particles have ability to prevent the adhesion between the two mating surfaces under unlubricated conditions. Ebdon (Ref 21) has reported that the low coefficient of friction of Ni-P-PTFE is due to the transfer of PTFE to the counter face material as compared to plain Ni-P (Ref 25), in both as-plated and heat-treated (400 °C/h) conditions. Ni-P-inorganic fullerene-WS2 (nanoparticles) deposit shows increased wear resistance property under oil-lubricated conditions as compared to Ni-P and Ni-P-graphite deposits (Ref 23). This is due to their ability to work as spacers between the asperities of two metal surfaces (Ref 21, 23) in contact.
For abrasive wear resistance properties, Ni-P-SiC coating proved to be the most cost-effective and best-performing combination (Ref 13, 14). As the incorporation level of SiC increases wear resistance properties also increase. As-plated Ni-P-hexagonal boron nitride (BN(h)) shows higher wear resistance than heat-treated (at 400 °C for 1 h) Ni-P coating (Ref 24). However, wear resistance properties nearly double when in the Ni-P matrix 33 vol.% BN(h) particles are incorporated (Ref 24).
In continuation of our earlier work (Ref 26, 27), owing to antibacterial activity and suitability under harsh processing condition of ZnO nanoparticles, in the present study co-deposition of synthesized ZnO nanoparticles into the Ni-P matrix on mild steel substrate by electroless technique was carried out. ZnO nanoparticles are synthesized by autocombustion method. For future application of Ni-P-ZnO nanocomposite coatings as antibacterial metallic coating their characterization, microhardness and corrosion resistance properties were carried out.
Experiment
Materials and Method
The electroless coating was carried out on mild steel (MS) sample, and the composition of mild steel sample is as follow:
Element | C | Si | Mn | P | S | Cu |
|---|---|---|---|---|---|---|
wt.% | 0.083 | 0.034 | 0.527 | 0.007 | 0.023 | 0.064 |
The mild steel (MS) sample of size (2 cm × 2 cm × 1.5 mm) was degreased with acetone and then cleaned with 10% aqueous NaOH solution to remove any greasy matter and corrosion product and then rinsed with deionized water. This is followed by sensitization with 1% SnCl2 (Loba Chemicals) and then activation with 0.01% PdCl2 (Merck) for ~1 min, the rinsing of sample is also carried out in between the steps. During sensitization process, Sn ions were deposited onto the sample surface to which Pd ions were deposited in activation step to provide nucleation/catalytic sites to facilitate the deposition process. Following bath composition and operating conditions were used for preparing Ni-P and Ni-P-ZnO coatings: NiSO4.6H2O (Sigma-Aldrich, 25 g/L), C6H5Na3O7.2H2O (Merck, 65 g/L), NaH2PO2.H2O (Loba chemicals, 20 g/L), ZnO (as-prepared, 10 g/L) at 90 ± 2 °C and pH 9.0 ± 0.2 maintained by using ammonia solution (NH4OH, Merck) along with buffer solution of (NH4)2SO4 (Sd-fine, 30 g/L) and NH4Cl (Merck, 30 g/L). The chemicals were used in the as-received form without any purification.
Characterization Techniques Used
The mechanical milling of the samples was carried out on High Energy Ball Mill (FRITSCH Model-P-7) for which zars and balls made up of zirconia were used. Thermal analysis of the milled ZnO powder was carried out using TG/DTA (PerkinElmer Diamond) in air at a heating rate of 10 °C/min. The structure of ZnO powder, Ni-P and Ni-P-ZnO coatings was studied by the powder x-ray diffractometer (XRD, D8 Bruker AXS Diffractometer) with CuKα radiation (γ = 0.1541 nm) and a monochromator 50 kV and 300 mA with the scanning rate and step being 2°/min and 0.02°, respectively. The surface morphology of the powder and coatings was examined by a Field Emission Scanning Electron Microscopy (FESEM, Quanta FEI-200) and a Scanning Electron Microscope (SEM, ZEISS EVO-18). The qualitative elemental analysis of the coatings was carried out using an Energy-Dispersive Analysis of x-rays (EDAX, PENTA FET Precision) an attachment of the FESEM equipment. In addition, Atomic Absorption Spectroscopy (Hitachi) was used to determine the elemental concentrations of Zn and Ni for which a set of standards (known concentration of each element) was prepared to calibrate the instrument before the actual measurements were taken. Crystallite average size of the powder was calculated by using Scherrer formula (d = 0.9λ/βcosθ) where D hkl is the crystallite size perpendicular to (hkl) plane, λ (nm) is the x-ray wave length used (i.e., λ = 1.514 Å), β (rad) is the width of the diffraction peak, and θ is the peak position. The full width at half maxima (FWHM) is used for β. The particle size of the synthesized ZnO powder was also confirmed by a transmission electron microscopy (TEM, TECNAI G2-20). The surface topography of the electroless coated samples was observed by atomic force microscopy (AFM-NTEGRA, TS150), and their histogram reveals the surface roughness (Ra) of the coated samples.
The corrosion behavior of the Ni-P/Ni-P-ZnO composite coatings was studied by potentiodynamic polarization tests/Tafel electrochemical measurements by Voltamaster 40 potentiostat, computer controlled with commercial software for the obtained data treatment (Ref 28). A three-electrode electrochemical cell configuration was employed having a saturated calomel electrode (SCE) as reference, and graphite as counter electrode, while the coated specimens were used as the working electrode. The test samples were exposed to aqueous solution of 3.5 wt.% NaCl at room temperature, and their linear polarization curves have been recorded at a constant scan rate of 1 mV/s, in the range of −0.25 to 1.4 V versus open circuit potential.
Results and Discussion
Synthesis of ZnO Nanoparticles
0.1 M solutions of zinc nitratehexahydrate (Zn(NO3)2·6H2O, Sigma-Aldrich) and 0.1 M citric acid monohydrate (C6H8O7·H2O, Sigma-Aldrich) were prepared by dissolving in deionized water (Ref 29). 0.1 M solution of Zn(NO3)2 and citric acid were mixed in 1:1 molar ratio with continuous stirring for 30 min at room temperature. Then the resulting homogeneous solution was heated at 80 °C with continuous stirring for 5-6 h (or depending on the volume of reaction mixture) till the reaction mixture is transformed into gel. After gel was formed and dried, it burnt in a self-propagating combustion manner until all gels were completely burnt (see Fig. 1) to form a fluffy, loose powder according to the chemical reaction 1.

SEM-EDAX of (a) as-synthesized ZnO powder, and (b) ZnO powder after 6 h ball-milling
Thermal analysis of the resulting powder is shown in Fig. 2 in which an exothermic peak is observed at 500 °C, suggesting a phase transformation. Therefore, the as-prepared powder was calcined at 500 °C for 3 h. The heat-treated (HT) powder was mechanically milled with 3:1 ball to powder ratio in a high energy ball-mill at 200 rpm for 6 h, at every 15-h interval, in air atmosphere (Fig. 1b). TEM image along with SADE pattern of the HT-mechanically milled powder is shown in (Fig. 3).

TG-DTA curve of as-prepared ZnO powder

TEM micrograph and SADE pattern of HT-mechanically milled, ZnO powder
Thermal Gravimetric Analysis of Synthesized ZnO Powder
In the TG curve, around ~3% wt loss is observed at 100-170 °C, indicating evaporation of moisture content or dehydration of the sample. An exothermic peak ~188 °C in DTA curve is observed as reported by Roy (Ref 29). This may be due to the autocatalytic anionic oxidation-reduction reaction of nitrates with citric acid, which initiate the formation of ZnO nanopowder by solid-state diffusion process. A peak observed at 504 °C (thermocouple e.m.f.: 152.6 µV; enthalpy: -5.08 J/mg) in DTA curve (Fig. 2) is exothermic as suggested by the minus sign of the enthalpy. A significant weight loss observed in the temperature range 400 °C to 575 °C in the corresponding TG curve, indicates some structural or phase change, corresponding to this weight loss step a peak observed at 507 °C (rate of wt loss, 0.63 mg/min) in DTG curves (Fig. 2). No further wt. loss is observed, which demonstrates that the decomposition of the precursor does not happen above this temperature and the stable residues may be ascribed as ZnO nanoparticles, which is confirmed by XRD (Fig. 4a). Hence, 500 °C is chosen as calcination temperature for the precursor ZnO powder. The ZnO powder used in Ni-P-ZnO coating was calcined at 500 °C for 3 h in air atmosphere and then mechanically milled for 6 h at every 15 min interval at the speed of 200 rpm. The sample thus obtained is analyzed by TEM, and from the micrographs (Fig. 3) it is observed that the prepared ZnO particles has spherical shape and crystalline nature.

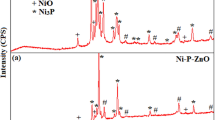
XRD spectra of (a) ZnO powder and (b) Ni-P and Ni-P-ZnO as-coated and heat-treated samples
Deposition of Ni-P/Ni-P-ZnO by Electroless Technique
The deposition process was carried out for 1 h, at constant temperature (at 90 ± 2 °C). At first, a Ni-P layer was deposited for 15 min (to prevent any porosity in the coating), and then ZnO nanoparticles (~40 nm size, determined by TEM) were introduced with constant stirring into the same bath for subsequent 45 min for co-deposition of ZnO into the Ni-P matrix (Fig. 4a). For comparison, plain Ni-P deposit (Fig. 4b) without ZnO particles was also prepared. After plating, the samples were taken out, rinsed with deionized water, then air-dried and stored in vacuum desiccators. Heat treatment of Ni-P and Ni-P-ZnO coatings was carried out in argon atmosphere at 400 °C for 1 h (Fig. 4c, d). On heat treatment of the coating, a phase transformation from microcrystalline to crystalline phase occurs because of the precipitation of Ni3P, which improves the adhesion of the coating to the substrate (Ref 30) and improve the properties w.r.t. as-coated sample.
XRD Analyses of Powder ZnO, Ni-P and Ni-P-ZnO Composite Coatings Samples
The XRD spectra of as-synthesized and heat-treated (at 500 °C for 3 h) followed by 6-h ball-milling, ZnO powder are shown in Fig. 4a. XRD spectra of as-synthesized, heat-treated (HT) and HT-mechanically milled ZnO powder are almost the same (except for the peaks intensity) and are in good agreement with the JCPDS file of ZnO (JCPDS card No. 00-036-1451, Fig. 4a, b) indicating a single hexagonal ZnO phase with P63mc structure. The sharp and intense peaks of ZnO HT-mechanically milled sample indicate that the sample has a crystalline structure. However, for mechanically milled sample, intensity of the peaks is lessen along with broadening of peaks, which clearly indicates the reduction of particle size (JCPDS card No. 01-080-0074, Fig. 4c). The XRD peaks for (100), (002) and (101) planes indicate the formation of phase pure wurtzite structure of ZnO. The size of HT-mechanically milled ZnO particles is determined by TEM (Fig. 3b) which is in the range of 50-100 nm. The crystallite average size of the ZnO powder calculated by using Scherrer formula by taking FWHM for (101) as highest intensity peak is in 50-70 nm range, which is in good agreement with TEM results.
XRD spectra of as-coated and heat-treated Ni-P and Ni-P-ZnO coatings are shown in Fig. 4(b). XRD spectrum of the as-plated Ni-P deposits is characterized by diffraction Ni(111), which corresponds to microcrystalline phase of the alloy [JCPDS card no. 00- 003-1044], and apart from this a broad peak of Fe corresponding to substrate was also observed. The microcrystalline nature of the coating is also justified by the presence of 6-8 wt.% of P contain (Table 1) observed by EDAX. The peak of Fe may arise from the mild steel substrate. The corresponding spectra of as-plated Ni-P-ZnO coatings also revealed the microcrystalline structure of Ni-P matrix along with the small characteristic diffraction peaks of ZnO particles. Small peaks of ZnO particles may be due to the presence of low concentration of ZnO particles in the composite coatings. Heat treatment at 400 °C for 1 h revealed a decrease in the line broadening and intensity of the peak for pure and composite coatings increases, which indicates crystallization of Ni-P matrix, i.e., the transformation of microcrystalline nickel and NixPy phases into crystalline Ni, Ni3P and ZnO phase (JCPDS Ref. No. 01-074-1384).
Morphology of ZnO Nanopowder and Ni-P/Ni-P-ZnO Coatings
FESEM micrographs of ZnO nanopowder, as-synthesized and heat-treated followed by ball-milling (HT ball-milled) are shown in Fig. 1(a-b). SEM micrographs suggest fluffy and porous morphology for as-synthesized ZnO powder. However, for HT ball-milled sample, an agglomeration of spherical shape particles is observed (Fig. 1b). The size observed for the calcined sample is in 50-100 nm range (Fig. 3b). The surface morphologies of as-coated and heat-treated Ni-P and Ni-P-ZnO coatings are shown in Fig. 5(a-d). After heat treatment, no significant change in topography of the coatings is observed. Microstructure of Ni-P coating showed the typical spherical globular homogeneous structure, but the morphology of composite Ni-P-ZnO coating is different from Ni-P. In Ni-P-ZnO, ZnO particles are well dispersed in Ni-P matrix but at some places agglomeration of ZnO particles also seems that may be due to the deposition of excess ZnO particles on the surface of the coating. The obtained composite layers had the spherical nodular structure. The higher magnified images show the embedded ZnO grains into the Ni-P matrix. The approximate stoichiometric ratio of elemental analysis (wt.%) determined by EDAX is given in Table 1. The concentration of Zn in the coating samples, prepared by scratching the coating and dissolved in HNO3 solution, was also determined by Atomic Absorption Spectroscopy (AAS), and the value obtained in ppm was converted into atomic wt.% is ~7.5% with reference to the Zn(NO3)2, standard solutions. The discrepancy in the EDAX and AAS results may be due to the determination of localized regions in EDAX, whereas the coating was scratched and dissolved in HNO3 solution to determine the Zn concentration. To verify uniform distribution of ZnO particles on the coated surface of composite Ni-P-ZnO (HT) sample, the elemental mapping studies through line spectrum were carried out (Fig. 6). The cross-sectional image with line scan analysis along with surface morphology and EDAX analysis is shown in Fig. 5d, and elemental line spectra are shown in Fig. 6. The mapping analysis indicates the presence of Ni, P and Zn elements in the coating and Fe of the substrate. The uniform distribution of Ni, P and Zn elements is seen in the entire matrix. Elemental mapping for oxygen was not carried out but its presence is clearly seen from EDAX analysis (Fig. 6b). Therefore, the uniform distribution of Zn in the analyzed portion of the coating indicates the entrapment of Zn in the form of ZnO particles throughout the coating and therefore, reveals the formation of uniform Ni-P-ZnO coating. On the other hand, in the present study the elemental (% wt, Table 1) analysis measured by EDAX reveals a decrease in concentration of Ni %, whereas P % remains almost same with unusual increase of % Fe in matrix on inclusion of ZnO (10 g/L). The results in our case look different from the results reported by Valova et al. (Ref 31) and Bouanani et al. (Ref 32). For Ni-Zn-P coating, i.e., on inclusion of Zn the % P content decreases into the coating, and Zn works as an inhibitor for the deposition process. But our results follow some similarity as observed by Veeraraghavan et al. (Ref 33), i.e., % Ni decreases and P remains almost same. It was reported (Ref 34-36) that the elemental distribution in the coating affects the coating properties and forms an interdiffusional layer. The presence of significant amount and uniform distribution of Fe revealed by EDAX and line spectrum analysis, respectively, may be attributed to the diffusion of coating elements toward interface of coating and mild steel substrate.

FESEM micrographs of (a) as-coated Ni-P, (b) HT Ni-P, (c) as-coated Ni-P-ZnO, (d) HT Ni-P-ZnO samples

X-ray mapping line spectra of heat-treated Ni-P-ZnO coating
AFM Topography of Coatings Samples
AFM images and surface profile of heat-treated Ni-P and as-coated and heat-treated Ni-P-ZnO deposits are shown in Fig. 7(a-c). The as-coated Ni-P AFM image was reported in (Ref 27). AFM micrographs of plain electroless Ni-P deposit (Fig. 7a) show a number of fine nodules throughout the surface (Ref 35). This may be explained by considering that the homogeneity of the surface is affected by the presence of ZnO in Ni-P layer and increases the number of boundaries between Ni and other particles of the matrix. It seems nodule size has increased with incorporation of ZnO particles in the Ni-P matrix as shown in Fig. 7(b). The ZnO grains embedded into the Ni-P matrix become finer on heat treatment and distinctly enlarge the surface development of the composite Ni-P-ZnO layer as compared to Ni-P. This fact is also confirmed by the higher average roughness value of initial deposition center and spot sizes, i.e., 23.54 nm for Ni-P-ZnO (HT) and 18.43 for Ni-P (HT) observed by AFM. Diameter of the very big nodule of ~40 µm is seen in composite coating. Uniform distribution of ZnO particle incorporation is seen throughout the surface of the coatings. Such a uniform distribution of the incorporated second phase particles clearly indicate a uniform supply of sufficient concentration of these particles at the coating-solution interface for incorporation. After heat treatment, the size of the nodular-like structure (Fig. 7c) decreases and the surface appears smoother with few nodules present on it. However, on heat treatment, the size of the grains increases that may be due to the diffusion of smaller grains into bigger ones to form diffused-like morphology with reduced surface roughness as observed from the curves.

AFM micrographs of (a) HT Ni-P, (b) as-coated Ni-P-ZnO, (c) HT Ni-P-ZnO samples
Microhardness Study of Ni-P/Ni-P-ZnO Coatings
Microhardness (VHN) of Ni-P and Ni-P-ZnO nanocomposite coatings in “as-coated” and HT conditions were determined using microhardness tester (402 MVD, Wilson Instruments) with dwelling time of 15 s under a 10 gf load. The microhardness of the substrate, Ni-P coated sample and Ni-P-ZnO coated samples, is given in Table 2. The microhardness of electroless Ni-P composite coatings increases with the incorporation of ceramic hard particles, whereas with soft particles, the microhardness tends to decrease. The level of incorporation of particles, the phosphorous content of the matrix and heat treatment determines the microhardness of coating (Ref 37). The results of the microhardness suggest that the addition of ZnO nanoparticles in the coating does not contribute significantly to the microhardness of the sample as ZnO nano particles are soft metal oxide as well as on further increasing the concentration of the ZnO particles the microhardness value decreases.
Corrosion Resistance study of Ni-P/Ni-P-ZnO Coatings
Corrosion is a surface phenomenon which occurs on the active surface of the substrate, which serves as a working electrode. The potential of the electrode-electrolyte interface can be measured using a reference electrode (saturated calomel electrode), and the nobleness of the coating surface is measured by open circuit (O.C.P.). The polarization curves of heat-treated electroless Ni-P and Ni-P-ZnO coatings in 3.5% NaCl solutions are shown in Fig. 8(a-b). From the graphs, the O.C.P. value observed for heat-treated Ni-P and Ni-P-ZnO coatings is -0.168 V and -0.152 V versus SCE, respectively. Shibli et al. (Ref 38, 39) have studied the corrosion resistant property of Ni-P-ZnO coatings and have reported the O.C.P. values -0.170 V and -0.163 V versus SCE for Ni-P and Ni-P-ZnO (0.5 g/L), respectively. In the present study, slightly nobler value is observed for Ni-P-ZnO coating which suggests improved corrosion resistance behavior of the coating. This improved corrosion resistant behavior of Ni-P-ZnO coating, either may be due to incorporation of large number of ZnO nanoparticles in the coating, which results a decrease in the electrochemically active area of the coating or the changed experimental conditions may be responsible for the observed shift in Ecorr values, e.g., inclusion of ZnO nanoparticles of size 20 nm for varied concentration viz. 0.1–2.0 g/L into the bath solution, for 3-h deposition time (Ref 38, 39). However, in the present study, 10 g/L ZnO nanoparticles of ~40 nm were added to the bath for 1-h deposition time with different hypophosphite concentration, and the conditions were optimized elsewhere (Ref 40). It was observed in (Ref 38, 39) that the thickness and hardness value of the coatings were increased and attains maxima for 0.5 g/L of nanoZnO and on further addition, i.e., up to 2.0 g/L the thickness as well as the hardness values is lesser than Ni-P-ZnO (0.5 g/L). The O.C.P. and deposition potential (Ed) reported in (Ref 39) for 0.5 and 2.0 g/L ZnO, are close to each other but along with other coating parameters 0.5 g/L ZnO was chosen as optimum concentration. Allahkaram et al. (Ref 37) has reported that the maximum amount of ZnO nanoparticles were incorporated into the coating from bath containing 2 g/L of ZnO and beyond 2 g/L the EL bath decomposes due to large amount of ZnO, which is responsible for increased surface area, i.e., excessive to loading factor. It was also reported (Ref 37) that Ni-P-ZnO coating shows a higher Rct (charge transfer resistance) as compared to Ni-P coating which may be due to amorphous character and more amount of nanoparticles on the surface decrease the electrochemically active area and hence increases the corrosion resistance. In our case, it was found that the deposition rate and hardness value attain maxima at 5 g/L ZnO (Ref 40) so the results obtained in our case are in partial agreement with the results reported elsewhere (Ref 37-39). The presence of higher % wt of Fe in case of Ni-P-ZnO heat-treated electroless coating matrix is unidentified for the authors. Hamdy et al. (Ref 41) have studied electroless Ni-P-Al2O3 nanocoatings and reported that heat treatment does not improve the localized corrosion resistance and the shifts of Epit and Ecorr to the nobler values may be due to the formation of a barrier coating layer for diffusion of oxygen to the metal surface and hence delaying but not preventing corrosion.

Electrochemical polarization curves for (a) HT Ni-P and (b) HT Ni-P-ZnO measured in 3.5 wt.% NaCl solution
The SEM micrographs of the specimens after corrosion test were also recorded and are shown in Fig. 9(a-b). As it was reported (Ref 42) that electroless Ni-P is a barrier coating, protecting the substrate from corrosive environments. The black spots appear at the surface of Ni-P coating (Fig. 9a) may be due to the penetration of chloride ions to the barrier layer of Ni-P alloy matrix or during surface preparation prior to the SEM analysis. Valova et al. (Ref 31) reported that the localization of zinc with P occurs primarily at the grain boundaries (Ref 31). In our case of EL Ni-P-ZnO nanocomposite coatings, black spots are also seen in the grain boundaries, it may be due to segregation of the nanoparticles followed by displacement of the ZnO particles during corrosion analysis. The spots of larger size revealed removal of large number of particles, and the smaller spots are developed by the displacement or segregation of a few numbers of the particles.

FESEM micrographs after corrosion test (a) HT Ni-P (c), (b) HT Ni-P-ZnO samples
Conclusion
A bright grayish and uniform nanocomposite coating of Ni-P-ZnO on mild steel substrates was obtained to improve the crystallinity of the coating. The electroless Ni-P-ZnO coating exhibits good adherence on the mild steel substrate. For the “as-coated” Ni-P-ZnO coating, the results (micrographs and line spectrum) of chemical composition obtained by FESEM-EDAX suggest that the particles of ZnO are uniformly co-deposited in the Ni-P matrix on the substrate surface with some excessive deposition of ZnO particles on the surface of the coating. The AFM micrographs reveal that the nodule size of the coating is increased on increasing the percentage of incorporation of second phase particle in the deposit. It is observed from the XRD spectrum of heat-treated sample that apart from high intensity Ni(1 1 1) peak, the peaks of Ni(2 0 0) and (2 2 0) are also present. Incorporation of second phase particles in the Ni-P deposit showed that there is a marginal variation in exothermic peak temperature compared to plain Ni-P deposit. A slight improvement in microhardness values for Ni-P-ZnO composite coatings is observed. The microhardness value is improved on incorporation of nanosize ZnO particles than the microsize particles into the Ni-P matrix, i.e., the microhardness value for Ni-P-ZnOnanosize (HT) is 590 and for Ni-P-ZnOmicrosize (HT) is 410 as determined from the previous study (Ref 27). In 3.5% NaCl solution, the corrosion resistance property of electroless Ni-P coating is slightly improved by the incorporation of ZnO particles.
References
A. Brenner and G. Riddell, Res. Natl. US Bur. Stand., 1946, 37, p 31
A. Brenner, D.E. Couch, and E.K. Williams, J. Res. Nat. Bur. Stand., 1950, 44, p 109
R.C. Agarwala and V. Agarwala, Sadhana, 2003, 28, p 475–493
R.C. Agarwala, Ph.D. thesis, Department of Metallurgical and Materials Engineering, University of Roorkee (presently IIT, Roorkee), (1987)
P.K. Datta, P.B. Bedingfield, D.B. Lewis and P.B. Wells, ‘Structure and phase changes accompanying treatment of electroless Ni-B alloy coating, Conf. Proc. 2nd Int. Electroless Nickel Conference Solihull, 1991, p 139–153
A. Srivastava, S. Mohan, V. Agarwala, and R.C. Agarwala, Z. Metallkd., 1992, 83, p 251–253
K.H. Krishnan, J. Praveen, M. Ganesan, P.M. Kavimani, S. John, and K.N. Srinivasan, Mater. Perform., 2006, 45, p 36–39
J.N. Balaraju and K.S. Rajam, Surf. Coat. Technol., 2005, 195, p 154–161
D.H. Kim, K. Aoki, and O. Takano, J. Electrochem. Soc., 1995, 142, p 3763–3767
L. Wang, L. Zhao, B. Zhang, S. Liao, Y. OnYang, and W. Hu, Z. Metallkd., 1997, 88, p 945–948
S.B Sharma, Ph.D. Thesis. ‘Synthesis and Tribological Characterization of Ni-P Based Electroless Composite Coatings’. IIT Roorkee, India, 2002
J. Sudagar, J. Lian, and W. Sha, J. Alloys Compd., 2013, 571, p 183–204
I. Apachitei, F.D. Tichelaar, J. Duszczyk, and L. Katgerman, Surf. Coat. Technol., 2002, 149, p 263–278
G. Jiaqiang, L. Lei, W. Yating, S. Bin, and H. Wenbin, Surf. Coat. Technol., 2006, 200, p 5836–5842
Y.S. Huang, X.T. Zeng, I. Annergren, and F.M. Liu, Surf. Coat. Technol., 2003, 167, p 207–211
D. Dong, X.H. Chen, W.T. Xiao, G.B. Yang, and P.Y. Zhang, Appl. Surf. Sci., 2009, 255, p 7051–7055
T. Rabizadeh and S.R. Allahkaram, Mater. Des., 2011, 32, p 133–138
W. Chen, W. Gao, and Y. He, Surf. Coat. Technol., 2010, 204, p 2493–2498
S.M. Moonir-Vaghefi, A. Saatchi, and J. Hejazi, Met. Finish., 1997, 95, p 46–52
M.D. Ger and B.J. Hwang, Mater. Chem. Phys., 2002, 76, p 38–45
P.R. Ebdon, Plat. Surf. Finish., 1988, 75, p 65–68
A. Ramalho and J.C. Miranda, Wear, 2005, 259, p 828–834
W.X. Chen, J.P. Tu, Z.D. Xu, R. Tenne, R. Rosenstveig, W.L. Chen, and H.Y. Gan, Adv. Eng. Mater., 2002, 4, p 686–690
O.A. Leon, M.H. Staia, and H.E. Hintermann, Surf. Coat. Technol., 2005, 200, p 1825–1829
N. Feldstein, G.O. Mallory and J.B. Hajdu, ‘Electroless Plating: Fundamentals and Applications’, American Electroplaters and Surface Finishers Society, Orlando, Florida,(Eds.), 1990, p 269.
S. Sharma, C.K. Saini, S. Sharma, and V. Agarwala, J. Mater. Environ. Sci., 2014, 5(5), p 1667–1670
S. Sharma, S. Sharma, P. Agarwala, R. Garg, and P. Gopinath, Adv. Mater. Res., 2012, 585, p 512–516
T. Rabizadeh, S.R. Allahkaram, and A. Zarebidaki, Mater. Des., 2010, 31, p 3174–3179
P.K. Roy, J. Mater. Sci. Res., 2012, 1, p 28–34
A. Zoikis-Karathanasis, E.A. Pavlatou, and N. Spyrellis, Electrochim. Acta, 2009, 54, p 2563–2570
E. Valova, I. Georgiev, S. Armyanov, J.L. Delplancke, B. Tachev, T. Tsa-Cheva, and J. Dille, J. Electrochem. Soc., 2001, 148(4), p 266
M. Bouanani, F. Cherkaoui, R. Fratesi, G. Roventi, and G. Barucca, J. Appl. Electrochem., 1999, 29, p 637
B. Veeraraghavan, B. Haran, S.P. Kumaraguru, and B. Popov, J. Electrochem. Soc., 2003, 150(4), p B131–B139
K. Zielinska, A. Stankiewicz, and I. Szczygiel, J. Colloid Interface Sci., 2012, 377, p 362–367
J.N. Balaraju, C. Anandan, and K.S. Rajam, Appl. Surf. Sci., 2005, 250, p 88–97
J.N. Balaraju, T.S.N. Sankara Narayanan and S.K. Seshadri, J. Appl. Electrochem., 2003, 33, p 807–816
S.R. Allahkaram, R. Faezi Alivand, and M.S. Bakhsh, Iran. J. Mater. Sci. Eng., 2013, 10, p 11–17
S.M.A. Shibli, B. Jabeera, and R.I. Anupama, Surf. Coat. Technol., 2006, 200, p 3903
S.M.A. Shibli, B. Jabeera, and R.I. Anupama, Appl. Surf. Sci., 2006, 253, p 1644–1648
Sarika, Ph.D. Thesis, Development of Electroless Ni-P-ZnO Nanocomposite Coatings, Graphic Era University, 2015
A.S. Hamdy, M.A. Shoeib, H. Hady, and O.F. Abdel, Salam, Surf. Coat. Technol., 2007, 202, p 162–171
A. Babanejhad, M. Hashemi, Y. Rahmatallahpur, and ShA Nozad, Bull. Mater. Sci., 2012, 35, p 561–566
Acknowledgments
Authors acknowledge Uttarakhand State Biotechnology Department (USBD), Haldwani, for financial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sharma, S., Sharma, S., Sharma, A. et al. Co-deposition of Synthesized ZnO Nanoparticles into Ni-P Matrix Using Electroless Technique and Their Corrosion Study. J. of Materi Eng and Perform 25, 4383–4393 (2016). https://doi.org/10.1007/s11665-016-2292-0
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11665-016-2292-0