
Abstract
Reliable, low-cost electrocatalysts of La and Ni co-doped LiMn2O4 for energy storage applications were successfully synthesized via the sol-gel method. Herein, the effect of La3+ and Ni2+ co-doping in Mn3+ of LiMn2−x(LaNi)xO4 (x = 0, 0.01, 0.02, 0.03, 0.04 and 0.05) samples were studied. X-ray diffraction (XRD) results for structural characterization established the cubic spinel structure of the materials. Microstructural studies of the synthesized materials via field emission scanning electron microscopy (FE-SEM) revealed that the particle size was reduced with La3+and Ni2+ doping. The electrochemical properties that were investigated in the voltage range of 0.2–1.2 V showed improved structural stability, and therefore Jahn–Teller distortion was effectively suppressed. Cyclic voltammetry (CV) revealed that a 4% substitution of (La and Ni) in Mn3+ exhibited improved lithium-ion reversibility. Electrochemical impedance spectroscopy (EIS) showed an improved charge transfer resistance of 41 Ω for (LaNi) 0.04 doping over other doping concentrations and pure LiMn2O4. Charge-discharge studies revealed that the initial discharge capacity of LiMn1.92(LaNi)0.04O4 is 132.1 mAh/g in aqueous Li2SO4 electrolyte is improved over other doping concentrations and pristine LiMn2O4.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the immense demand for high-energy-density storage systems, replacing conventional graphite anodes in Li-ion batteries (LIBs) offers a considerable opportunity to develop improved batteries for future applications. LiMn2O4 (LMO), the most promising intercalation material, has received significant attention as a cathode in LIBs. This spinal material offers advantages including environmental soundness, low cost, and good thermal behavior, without the need for expensive safety devices, which distinguish it from LiCoO2 and LiNiO2.1,2,3,4 However, limitations including capacity fading, poor high-rate capability, Mn dissolution at high temperature, and the Jahn–Teller effect are challenges that must be solved before its commercial use. To alleviate these issues, numerous researchers across the globe have developed various strategies such as synthesis methods, annealing atmospheres, iso- or aliovalent cations such as Li, Ni, Cu, Co, Al, and Cr,5,6,7,8 multiple cation substitution,9,10 and co-doping/dual doping.11,12 Among these methods, bication doping with rare-earth elements represents a possible method to address all these issues.13
Lanthanum doping has already shown some synergetic effects.14 Rare-earth element substitution has been proven to prevent the collapse of the spinel lattice by acting as a pillar for facilitating Li-ion mobility and providing faster intercalation/de-intercalation.15,16 Ni substitution is an emblematic method to advance the electrochemical activity of LiMn2O4.17,18,19 Ni substitution alone does not show any significant improvement, but when co-doped with other elements, like Mn, showed improved performance as a cathode material for a LIB. Therefore, Ni is chosen for this reason.
In this paper, we present a proof-of-concept demonstration of La and Ni co-doped LiMn2−x(LaNi)xO4 as a cathode material for a LIB application. We systematically study its structural, morphological, electrical, and electrochemical performance by doping at dopant concentrations of 1–5%. The electrochemical studies are carried out by fabricating a half cell in aqueous electrolytes.
Experimental
Synthesis
LiMn2−x(LaNi)xO4 (x = 0.01, 0.02, 0.03, 0.04, 0.05) was synthesized via a sol-gel process. Stoichiometric amounts of lithium acetate Li(CH3COO), manganese acetate Mn(CH3COO)2, lanthanum acetate La(CH3CO2)3·xH2O, and nickel acetate Ni(OCOCH3)2·4H2O (all chemicals of 99.99% purity, Aldrich) were used as precursor materials. The remaining process was as described in our previous paper for doped and pristine materials.20
Physical Characterization
The crystal structure, phase purity, and lattice parameters of the synthesized samples were obtained from X-ray diffraction (XRD) using a D8 Advance diffractometer (Bruker AXS) with Cu-Kα (λ = 1.54 Å) radiation at a scan rate of 2°/min in the range of 10°–80°. Microstructural and morphological studies were carried out by field emission scanning electron microscopy (FE-SEM; JEOL JSM-7600F).
Electrode Preparation and Electrochemical Measurements
An electrochemical analyzer (CHI 660E, CH Instruments, Inc., USA) was used to test cyclic voltammetry (CV), chronopotentiometry (CP), and electrochemical impedance spectroscopy (EIS) of LMO and doped materials in a prototype aqueous electrochemical cell. The cell (Pt//LiMn2O4) was designed using three electrodes immersed in a saturated Li2SO4 aqueous electrolyte. The working electrode was prepared by mixing LiMn2−x(LaNi)xO4, carbon black, and polyvinylidene fluoride (PVDF) in a ratio of 90:5:5. A slurry was formed by adding n-methyl pyrrolidone (NMP) dropwise. This slurry was coated on stainless steel foil of 2 mm thickness and heated for 12 h in a vacuum oven to remove NMP and any moisture in the material. Pt wire is used as a counter-electrode and Ag/AgCl as a reference electrode. The CV study was done at a scan rate of 0.5 mV/S in the potential range of 0.2–1.2 V. EIS data was taken in the frequency range of 10 MHz–100KHz. CP studies were carried out at a current density of 100 µA/cm2 in a potential range of 0.0–1.2 V.
Results and Discussion
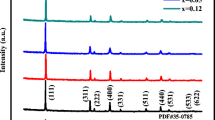
The crystallographic structure of as-prepared materials was studied using XRD. XRD analysis is the appropriate method to analyze small-scale microstructural phases and their structural properties.21, 22 The obtained XRD patterns of synthesized LiMn2−x(LaNi)xO4 materials are shown in Fig. 1. The majority of diffraction peaks perceived in the XRD pattern correspond to La3+ and Ni2+ doped in Mn3+/Mn4+ of 8a sites in LMO. The results depicted a cubic spinel assembly in the space group Fd3m, in good agreement with JCPDS (file no. 35-0782). Further, lattice parameters and crystallite sizes were calculated using Scherer's formula. The volume of the unit cell is depicted in Table I. It clearly shows that with concentration escalation, the lattice considerations reversibly decrease, leading to shrinkage of the crystalline cell. Shrinkage of the cell strongly suggests the reduction of Mn[La/Ni]-O bond length in Mn[La/Ni]O6 octahedrons and expansion of LiO4 tetrahedrons in the catalyst material's structure. The decrease in cell size corresponds to the strengthening of bonds, leading to enhanced electrocatalytic activity. Among all these doping concentrations, at higher doping concentrations (x = 0.05) some minor peaks were observed, which correspond to Mn2O3. This is due to the formation of minor oxides of La and Ni, which decompose completely into Mn2O3 in company with La(CH3CO2)3·xH2O during sintering in air.

XRD patterns of (a) LiMn2O4, (b) LiMn1.98(LaNi)0.01O4, (c) LiMn1.96(LaNi)0.02O4, (d) LiMn1.94(LaNi)0.03O4, e) LiMn1.92(LaNi)0.04O4, f) LiMn1.90(LaNi)0.05O4.
In the XRD data, broadening of the peaks is expected from three different sources: namely, instrumental, crystallite size and lattice strain. After calibration of a Si standard, we removed the instrumental effect using the Si standard background. Thereafter, the existing broadening is due to the crystallite size effect and the lattice strain present in the material.
Since the synthesized material is in the nano region (estimated using Debye–Scherer’s formula, and as already discussed above), additional broadening is present in the diffraction peaks from lattice strain. The preparation conditions probably introduce the lattice strain in the material, which leads to broadening of the peaks apart from the crystallite size effect. We can calculate the individual contributions of the crystallite size effect and lattice strain in the peak broadening. The existing peak widths (FWHM, full width at half maximum) can be written as
where
where λ is the wavelength of the x-ray used, θ is the Bragg angle, L is the average crystallite size, K is constant (0.94 for cubic crystals of uniform size and for other non-cubic crystals) and η is strain in the material. By plotting Br cosθ against sinθ, we obtained a straight line with slope of 0.3432, which equates to lattice strain present in the material, and the intercept of 0.0809 equates to Kλ/L. From this, we compared the crystallite size value L with the value calculated using Debye–Scherer’s formula, and they are in good agreement.
The exterior morphology of LMO and its doped materials examined by FE-SEM is depicted in Fig. 2a, b, c, d, e, and f. From this figure, it is observed that the particles are well shaped in a polyhedron structure (shown in inset) with a relatively smooth surface approximately 300 nm in size. A small particle is visible connecting larger particles, indicating that the crystallization process was complete. The EDS carried out for LMO and its doped derivatives are depicted in Fig. 3. EDS results for the sintered samples confirmed the presence of Mn, La, Ni, and O in all the samples. In addition, structural connectivity in these materials was studied by FTIR, and the spectra are shown in Fig. 4. The absorption bands in LMO between 620 cm−1 and 509 cm−1 were attributed to stretching modes of Mn-O bonds. In FTIR, bond strength is related to that bond's wave number23. In doped materials, the bond length of Mn[La/Ni]-O decreased with an increase in doping content, and it is minimum for (LaNi)0.04 doping. After doping, all of the peaks shifted to higher wave numbers, which shows that (LaNi)0.04 doping improved structural stability and enhanced the electrochemical properties of the material. The XRD results from Fig. 1 and CV results from Fig. 5 confirmed that (LaNi)0.04 substitution improves the properties of LMO.

FE-SEM micrographs of (a) LiMn2O4, (b) LiMn1.98(LaNi)0.01O4, (c) LiMn1.96(LaNi)0.02O4, (d) LiMn1.94(LaNi)0.03O4, (e) LiMn1.92(LaNi)0.04O4, and (f) LiMn1.90(LaNi)0.05O4.

EDS profiles of (a) LiMn2O4, (b) LiMn1.98(LaNi)0.01O4, (c) LiMn1.96(LaNi)0.02O4, (d) LiMn1.94(LaNi)0.03O4, (e) LiMn1.92(LaNi)0.04O4, and (f) LiMn1.90(LaNi)0.05O4.

FT-IR spectra of LMO and its derivatives.

Cyclic voltammograms of LiMn2O4, (a) LiMn1.98(LaNi)0.01O4, (b) LiMn1.96(LaNi)0.02O4, (c) LiMn1.94(LaNi)0.03O4, (d) LiMn1.92(LaNi)0.04O4, and (e) LiMn1.90(LaNi)0.05O4 for first and fifth cycles in saturated aqueous Li2SO4 electrolyte at a scan rate of 0.05 mV/s.
Electrochemical Performance of As-Prepared LMO and Doped LMO Materials
The electrochemical performance of LMO and La3+/Ni2+ doped LMO material were evaluated by CV in the cell potential range of 0.2 V to 1.2 V at a 0.05 mV/s scan rate and by CP. The CV curves of pristine and LiMn2−x(LaNi)xO4 materials for the first and fifth cycles are depicted in Fig. 5. Two pairs of well-resolved, sharp redox peaks appeared at 0.74 V, 0.89 V, and 0.81 V, 1.13 V for all materials in saturated aqueous Li2SO4 electrolyte. These peaks correspond to the conversion of Ni2+/Ni3+ and Ni3+/Ni4+. The potential variance amongst the oxidation and the reduction peaks (ΔEp) for the Ni2+/Ni3+ and Ni3+/Ni4+ redox processes was 0.095 V and 0.11 V, respectively. Small ΔEps values indicate fast Li+ extraction/insertion kinetics in the as-prepared materials. According to previous studies,24 the features of the obtained CVs indicate that the as-prepared LMO exhibited a mainly disordered structure. These results are in good arrangement with the redox potentials obtained for non-aqueous electrolytes.25,26,27,28 Among all doped materials, Fig. 5d LiMn1.92(LaNi)0.04O4 exhibited superior current density, fivefold higher than the other doped materials; this might be because of the good structural arrangement confirmed by the FE-SEM study. Variations in peak potential differences with different doping concentrations are explained in Fig. 6. As the doping concentration of La and Ni increases, the peak potential difference decreases, and the current response increases.29,30,31 As the structure became more stable with increasing La and Ni as described in the XRD result (x = 0.04), the dopant showed better electrochemical activity. Due to the formation of the secondary phase at higher doping concentration (shown in XRD), the bonds in Mn[La/Ni]O6 octahedrons may weaken and lead to a decrease in electrochemical activity. In all the samples, a matching current response was observed at the first and fifth cycles, indicating electrochemical stability. The second redox peaks in all of the samples showed an increase in current response showing higher charge-discharge capacity at the greater potential region and increased current response.

Peak potential comparison from cyclic voltammetry for LiMn2−x(LaNi)xO4.
EIS results through Nyquist AC impedance plots for LiMn2−x(LaNi)xO4 are shown in Fig. 7 for the first and fifth cycles.32 For dopant (x = 0.04), a lower Rct value (41 Ω) measured over the remaining doped and pristine samples indicated improved and faster Li-ion intercalation kinetics. The morphology of LiMn2−x(LaNi)xO4 is an important characteristic that affects the solid-state diffusion of Li+. Due to the other phase observed in (x = 0.05) concentration, doping led to a change in the distribution of Li+ and affected the electrochemical performance. These EIS results were consistent with the CV analysis. Variation of Z′ and Z′′ parts of impedance in the frequency range 10 MHz–100 KHz at different doping values are depicted in Fig. 8 (A and B). From figure A, low Z′ values observed for LiMn1.92(LaNi)0.04O4 show a decrease in Mn-La-Ni-O bond lengths, which in turn increase the Li+ conduction path. Z″ versus log F curves in figure B show no loss of materials even after the fifth cycle in each concentration. Z′ and Z″ curves merge at higher frequencies in both graphs, which indicates reduced space charge polarization with increasing frequency. (LaNi)0.02 doping shows low Z′ and Z″ values, indicating higher accumulation charges, which results in the improvement of electrochemical properties.

Nyquist plots of (a) LiMn2O4, ((b) LiMn1.98(LaNi)0.01O4, (c) LiMn1.96(LaNi)0.02O4, (d) LiMn1.94(LaNi)0.03O4, (e) LiMn1.92(LaNi)0.04O4, and (f) LiMn1.90(LaNi)0.05O4 for first and fifth cycles.

Variation of real part of impedance (top half); LMO-LiMn2O4, (a) LiMn1.98(LaNi)0.01O4, (b) LiMn1.96(LaNi)0.02O4, (c) LiMn1.94(LaNi)0.03O4, (d) LiMn1.92(LaNi)0.04O4, (e) LiMn1.90(LaNi)0.05O4 and imaginary parts of impedance (bottom half); LMO-LiMn2O4, (a) LiMn1.98(LaNi)0.01O4, (b) LiMn1.96(LaNi)0.02O4, (c) LiMn1.94(LaNi)0.03O4, (d) LiMn1.92(LaNi)0.04O4, (e) LiMn1.90(LaNi)0.05O4 with logF for first and fifth cycles.
Typical charge-discharge curves of LiMn2−x(LaNi)xO4 obtained for several cycles in the potential range of 0-1.2 V are depicted in Fig. 9. All the curves present two plateaus corresponding to lithium intercalation/de-intercalation and are consistent with the CVs.33,34,35 The figure shows that discharge capacity improved with La3+ and Ni2+ doping in Mn3+. The improved capacity with doping is attributed to shorter path lengths in which Li+ moves. La3+, Ni2+, and Mn3+ ions resided in the 16d site originally occupied by Mn3+/Mn4+ ions and caused Mn[La/Ni]O6 octahedrons to shrink, providing greater stability. Therefore, the electrochemical behavior of the doped derivatives was enhanced with increasing doping. The discharge capacity of the (x = 0.04) doped sample was 132.1 mAh/g, which is comparatively more than the discharge capacity of 113.2 mAh/g for pristine LMO. At a doping concentration of 0.05, due to the formation of the secondary phase, the bonds weakened and the discharge capacity suddenly decreased. These CP results showed a positive effect for the appropriate doping of La3+ and Ni2+ in Mn3+ of LMO.

Charge-discharge profiles of LiMn2O4 and its doped derivatives.
Conclusions
In this work, we successfully prepared a series of La-Ni doped LMO spinel materials by a sol-gel method and investigated their electronic charge transport properties and electrochemical performance. The doped samples preserve the spinel cubic structure except at higher doping concentrations, in which Mn2O3 oxides were observed. XRD results showed that La3+, and Ni2+ doping improved the stability of LMO. FE-SEM micrographs showed well-defined polyhedral morphology with particle sizes in the range of 200 nm, and the FE-SEM showed a relatively smooth surface. CV results showed two pairs of redox peaks with a decrease in peak potential differences at increasing doping concentration. 4% La3+ and Ni2+ doping showed improved electrochemical properties over other dopants. EIS curves showed an improved Li+ intercalation/de-intercalation process due to a decrease in Rct with increasing doping. Charge-discharge curves exhibited improved discharge capacity of 132.1 mAh/g for 4% La and Ni doping over other dopants and pure LMO. The enhanced electrochemical performance of LiMn1.92(LaNi)0.04O4 in Li2SO4 aqueous electrolyte suggests it can be used in energy storage devices.
References
N. Maragani and K. Vijaya Kumar, Rasayan J. Chem. 10, 1128 (2017).
X. Lv, S. Chen, C. Chen, L. Liu, F. Liu, and G. Qiu, Solid State Sci 31, 16 (2014).
J. Ramesh Babu, K. Ravindhranath, and K. Vijaya Kumar, Asian J. Chem. 29, 1049 (2017).
K. Sreekanth, T. Siddaiah, N.O. Gopal, N. Krishna Jyothi, K. Vijaya Kumar, and Ch. Ramu, J. Macromol. Sci. Part B Phys. 58, 860 (2019).
G. Maino, J. D’Haen, F. Mattelaer, C. Detavernier, A. Hardy, and M.K. Van Bael, J. Mater. Chem. A 4, 18457 (2016).
R. Thirunakaran, A. Sivashanmugam, S. Gopukumar, C.W. Dunnill, and D.H. Gregory, J. Mater. Process. Technol. 208, 520 (2008).
S. Mandal, R.M. Rojas, J.M. Amarilla, P. Calle, N.V. Kosova, V.F. Anufrienko, and J.M. Rojo, Chem. Mater. 14, 1598 (2002).
Y. Ein-Eli, R.C. Urian, W. Wen, and S. Mukerjee, Electrochim. Acta 50, 1931 (2005).
R.M. Rojas, J.M. Amarilla, L. Pascual, J.M. Rojo, D. Kovacheva, and K. Petrov, J. Power Sources 160, 529 (2006).
H. Goktepe, H. Sahan, S. Patat, and A. Ulgen, Ion 15, 233 (2009).
R. Thirunakaran, A. Sivashanmugam, S. Gopukumar, and R. Rajalakshmi, J. Power Sources 187, 565 (2009).
A. Ramakrishna, N. Murali, S.J. Margarette, T. Wegayehu Mammo, N. Krishna Jyothi, B. Sailaja, C.C. Sailaja Kumari, K. Samatha, and V. Veeraiah, Adv. Powder Technol 29, 2601 (2018).
H. Liu, Li. Song, and K. Zhang, Inorg. Mater. 41, 646 (2005).
D. Arumugam, G.P. Kalaignan, and P. Manisankar, Solid State Ion 179, 580 (2008).
H. Sun, D. Zhu, Y. Chen, C. Xu, L. Huang, and H. Yang, J. Solid State Electrochem. 16, 2979 (2012).
S. Maity, B. Das, R. Maity, N.P. Maity, K. Guha, and K.S. Rao, Sol. Energy 185, 439 (2019).
Y. Deng, J. Mou, H. Wu, L. Zhou, Q. Zheng, K.H. Lam, C. Xu, and D. Lin, ChemElectroChem 4, 1362 (2017).
Y.J. Wei, L.Y. Yan, C.Z. Wang, X.G. Xu, F. Wu, and G. Chen, J. Phys. Chem. B 108, 18547 (2004).
M. Kebede, N. Kunjuzwa, K. Ozoemena, and M.K. Mathe, J. Electrochem. Soc. 50, 1 (2013).
A. Venkateswara Rao, V. Veeraiah, A.V. Prasada Rao, B. Kishore Babu, and K. Vijaya Kumar, Ceram. Int. 44, 15116 (2018).
Y. Tomota, M. Ojima, S. Harjo, W. Gong, S. Sato, and T. Ungár, Mater. Sci. Eng. A 743, 32 (2018).
H.P.A. Ali, N. Tamura, and A.S. Budiman, IEEE Trans. Device Mater. Reliab. 18, 490 (2018).
R. Rathika, M. Kovendhan, D.P. Joseph, K. Vijayarangamuthu, A.S. Kumar, C. Venkateswaran, K. Asokan, and S.J. Jeyakumar, Nucl. Eng. Technol. 52, 2585 (2019).
J. Madhuri Sailaja, N. Murali, S.J. Margarette, N.K. Jyothi, K. Rajkumar, and V. Veeraiah, S. Afr. J. Chem. Eng. 26, 61 (2018).
R. Vemuri, G. Raju, M. Gnana Kiran, M.S.N.A. Prasad, E. Rajesh, G. Pavan Kumar, and N. Murali, Results Phys. 12, 947 (2019).
H.M. Wu, J.P. Tu, Y.F. Yuan, Y. Li, X.B. Zhao, and G.S. Cao, Mater. Sci. Eng. B 119, 75 (2005).
T. Doi, T. Yahiro, S. Okada, and J. Yamaki, Electrochim. Acta 53, 8064 (2008).
R. Ravindran, S. Ernest, P. Thomas, and S. Renganathan, Bull. Mater. Sci. 42, 1 (2019).
C.Y. Khoo, H. Liu, W.A. Sasangka, R.I. Made, N. Tamura, M. Kunz, A.S. Budiman, C.L. Gan, and C.V. Thompson, J. Mater. Sci. 51, 1864 (2016).
X. Liu and T.C. Weng, MRS Bull. 41, 466 (2016).
S.K. Tippabhotla, I. Radchenko, C.V. Stan, N. Tamura, and A.S. Budiman, J. Mater. Res. 34, 1622 (2019).
F.X. Wang, S.Y. Xiao, Y. Shi, L.L. Liu, Y.S. Zhu, Y.P. Wu, J.Z. Wang, and R. Holze, Electrochim. Acta 93, 301 (2013).
Y. Cui, Z. Yuan, W. Bao, Q. Zhuang, and Z. Sun, J. Appl. Electrochem. 42, 883 (2012).
C.-H. Lu and S.K. Saha, Mater. Sci. Eng. B 79, 247 (2001).
R. Samal, B. Dash, C.K. Sarangi, K. Sanjay, T. Subbaiah, G. Senanayake, and M. Minakshi, Nanomaterials 7, 356 (2017).
Acknowledgments
The authors gratefully acknowledge the management of the Koneru Lakshmaiah Education Foundation (KLEF) for their encouragement and support; Dr. A. Venkateswara Rao is thankful to DST for providing financial assistance DST-SERB, Young Scientist Project: File No. SB/FTP/ETA-0176/2014, and also the Department of Science and Technology (DST-SERB), Govt. of India, for the award of DST-FIST level-I (SR/FST/PS-I/2018/35) to the Department of Physics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors who contributed to this paper wish to confirm that there are no known conflicts of interest associated with this publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Madhu, M., Venkateswara Rao, A. & Mutyala, S. La and Ni Co-doping Effect in LiMn2O4 on Structural and Electrochemical Properties for Lithium-Ion Batteries. J. Electron. Mater. 50, 5141–5149 (2021). https://doi.org/10.1007/s11664-021-09037-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-021-09037-w