
Abstract
Silico-ferrite of calcium (SFC) is a key intermediate phase in the sintering process of fine iron ores, and SiO2 plays an important role in the formation of SFC. In this work, the crystal structure stability of SFC synthesized at 1473 K (1200 °C) has been determined by X-ray diffraction, field-emission scanning electron microscopy, and X-ray absorption spectra. Synthesis of SFC was carried out under air at 1473 K (1200 °C) by mixing different amounts of SiO2 with Fe2O3 and CaCO3. The results show that the maximum solid solubility of SiO2 in the crystal structure of SFC does not exceed 6.11 wt pct at 1473 K (1200 °C); under these conditions, Fe2O3 begins to appear. The process of Si solution is closely related to the presence of a Ca channel composed of Ca octahedron in the crystal structure of SFC based on the results from the measurements of Ca K-edge X-ray absorption spectra. Si mainly occupies the center positions of the upper and lower tetrahedron adjacent to Ca channel. The length of Ca-Ca bond in Ca channel increases with the increasing of Si content. The crystal structure stability of SFC may be related to the structure of the Ca channel.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The quality of iron ore sinter is largely determined by the phase proportion of the silico-ferrites of calcium and aluminum (SFCA) which is a low-melting point bonding phase formed during the sintering process.[1,2] The SFC phase was identified by Hamilton et al.[3], who recognized this as an alumina-free equivalent of SFCA with a compositional range located along the CaO·3Fe2O3 (CF3)—4CaO·3SiO2 (C4S3) pseudobinary. Since then, Pownceby et al.[4–7] have systematically studied the solid solution limits and thermal stability of SFC, and its alumina-doped equivalent SFCA, and also found that SFC forms a solid solution along a trend line between the theoretical end-members CF3 and C4S3. The maximum solid solution range has been reported to occur between compositions containing approximately 7.0 to 11.7 wt pct C4S3 component (about 3.12 and 5.21 wt pct SiO2), and the solution range is valid between 1333 K and 1513 K (1060 °C and 1240 °C). The reason for the limited SiO2 solubility in SFC in this range is not very clear at present.
The discovery of SFC is playing an important role in determining the composition of complex silico-ferrites of calcium and aluminum (SFCA-Ι and SFCA), the solid solution along the CF3-C4S3 pseudobinary is valid for SFCA-Ι and SFCA, as well as SFC, and Al3+ just directly substitutes for Fe3+.[8–11] Lister and Glasser[12] have found that CaO·3(Al2O3, Fe2O3) defined as “ternary phase” (TP) appeared in the Fe2O3-CaO-Al2O3 system and is stable between 973 K and 1723 K (700 °C and 1450 °C). Mumme et al.[13] subsequently studied this phase in detail and determined the crystal structure of Ca5.1Al9.3Fe 3+18.7 Fe 2+0.9 O48, which thus designated as SFCA-ΙΙ. Webster and Pownceby et al.[10,11] also found that TP appeared in the formation of SFCA. Al2O3 was confirmed to form solid solution with calcium ferrite by the substitution between Al3+ and Fe3+ before the formation of complex SFCA, but the crystal structure of Al2O3 contained calcium ferrite is different with that of SFCA. These findings proved that the formation of complex SFCA cannot take place without SiO2 participation. Research on the crystal structure of SFC provides the opportunity to obtain a deeper understanding of the structure of SFCA.
In a recent study on the structure and formation mechanism of SFC, Ding et al.[14] have found that Si is located on the center of the upper and lower tetrahedral structure near a Ca channel (formed by the stacking of Ca-Ca along c-axis, see Figure 1(b)). Ca replaces Fe in the center of octahedra adjacent to Si tetrahedra in the crystal structure of SFC (see Figure 1). There is a lack of experimental data of the structural changes taking place in SFC, and considering that the content of Si in SFC is relatively low, the Ca K-edge X-ray absorption spectra of SFC with different contents of SiO2 were determined. SFC samples were synthesized with varying SiO2 content by solid-state reaction at 1473 K (1200 °C), and the phase compositions of the samples were studied by X-ray diffraction (XRD) and field-emission scanning electron microscopy (FESEM).

Composition of (a) the octahedral and tetrahedral layers; and (b) the crystal structure of SFC. (The frames composed by the red dashed line represent the positions of Ca and Si solution in the structure of SFC, respectively)[14]
Experimental
Sample Preparation
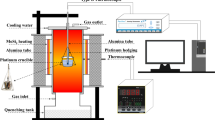
Fine-grained and dehydrated Fe2O3, SiO2, and CaCO3 powders (Sinopharm Chemical Reagent Co., Ltd., Beijing, China) were used as the starting materials. Considering the minimum content of SiO2 required for the formation of SFC and that the solution mechanism of Si mainly is 2Fe3+ = Ca2+ + Si4+,[5,10] SFC was synthesized by changing the amounts of Fe2O3, SiO2, and CaO in a certain range. The bulk compositions of samples are presented in Table I. Each group of samples was homogenized by grinding in anhydrous ethanol for approximately 20 minutes using an agate mortar, then the samples were put into a drying oven at 343 K (70 °C) and milled for approximately 10 minutes again after drying to ensure thorough homogenization.
The dried samples were pressed into 2 g pellets using a briquetting machine. Then, the pellets were sintered in platinum crucibles at 1473 K (1200 °C) for 12 hours in air. After that, the sintered samples were crushed, re-ground, pressed into pellets, and sintered at 1473 K (1200 °C) for another 12 hours to ensure that the thermodynamic equilibrium was reached. All sintering experiments were conducted in a box-type resistance furnace (muffle furnace). In addition, the temperature of synthesis experiment was monitored with a Pt90Rh10-Pt (type S) thermocouple positioned directly next to the sample; the thermocouple was calibrated against the melting point of gold, and the temperatures are considered accurate to ±3 °C to 4 °C.
Phase Determination [XRD, Field-Emission Scanning Electron Microscopy (FESEM)]
A part of each annealed sample was ground into a fine powder in an agate mortar and pestle and then sieved to produce a 50 μm powder for XRD analysis. XRD analysis was conducted using a Rigaku Ultima IV X-ray diffractometer (Rigaku Corporation, Tokyo, Japan), with Cu Kα used as the radiation source (40 kV, 400 mA) and a graphite monochromator in the diffraction beam path. The XRD data were collected using continuous scanning mode, of which the scanning speed was 10 deg/minute and the diffraction angle 2θ range from 10 deg to 90 deg.
The remaining part of the annealed samples was sectioned, polished after setting into an ethylenediamine-doped epoxy resin, and prepared for FESEM. FESEM was performed using a Zeiss Ultra 55 field-emission scanning electron microscope (CARL ZEISS Optical Instrument Co. Ltd., Jena, Germany), at an accelerating voltage of 15 kV. Energy-dispersive spectroscopy (EDS) was performed on this instrument using an Oxford X-Max 50 EDS detector (Oxford Instruments, Oxford, U.K.).
X-ray Absorption Spectra (XAS)
The remaining part of the powder samples was used for XAS measurements. The XAS experiments were performed at the beamline 4B7A of Beijing Synchrotron Radiation Facility (Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, China). Ca K-edge X-ray absorption near-edge structure (XANES) spectra were recorded under helium atmosphere and at the ambient temperature of 300 K (27 °C). The scanned energy range was between 3975 and 4185 eV, with the minimum energy step of 0.3 eV and the dwell time of 1 s per point. During the data collection, the storage ring current ranged between 420 and 620 mA. The data were collected in the total electron yield mode.
Results and Discussion
XRD Measurements
XRD results of the samples after solid-state reaction are shown in Figure 2. It can be seen that the main phase is SFC in each sample with the content range of SiO2 from 3.12 wt pct (S1) to 6.11 wt pct (S4). In addition, there are two small diffraction peaks apparent in the samples S1 (SiO2 = 3.12 wt pct) and S4 (SiO2 = 6.11 wt pct) with the 2θ angle near 33.5 deg (as shown by the red dashed line box in Figure 2), respectively.

XRD patterns of samples after solid-state reaction at 1473 K (1200 °C) for 24 h and comparison with the peak positions for SFC from Pownceby et al.[5] and Ding et al.,[14] respectively. S1 to S4 refer to the different compositions listed in Table I. The diffraction peaks in the red dashed line box do not belong to SFC
The diffraction peaks appeared at 2θ angle near 33.5 deg were identified through The International Centre for Diffraction Data database, to be CaFe2O4 (CF) and Fe2O3, respectively (see Figure 3). It can be considered that CaFe2O4 appeared in S1 (SiO2 = 3.12 wt pct) due to the relatively low amount of SiO2, and excess Fe2O3 reacted with CaO to form CaFe2O4 in the sample through solid-state reaction. The emergence of very small amount of Fe2O3 in S4 (SiO2 = 6.11 wt pct), in which the content of SiO2 is relatively high, can be regarded as exceeding the solid solubility of Si in the structure of SFC. The reason of this happened would be analyzed by Ca K-edge XANES in the following experiment.

XRD patterns of the second phases appeared in S1 (SiO2 = 3.12 wt pct) and S4 (SiO2 = 6.11 wt pct), respectively
FESEM Examination
Figure 4 shows the secondary electron images for the samples S1 (SiO2 = 3.12 wt pct), S2 (SiO2 = 4.01 wt pct), S3 (SiO2 = 4.90 wt pct), and S4 (SiO2 = 6.11 wt pct). It can be seen that the compositions of S2 and S3 are mainly SFC, and a small amount of Fe2O3 appears in S4. The results are consistent with the XRD results. Note, however, it is difficult to distinguish SFC and CF in S1 under the scanning electron microscope (the left part of S1 FESEM image in Figure 4(a)) even in backscattered mode due to similarities in composition. It was found that the pure phases of SFC and CF exhibit different changes after contact with hydrofluoric acid (of which the concentration of HF is 2 wt pct). SFC was completely dissolved, while CF showed no obvious change (the result of EDS shown in Figure 5(a)). This provides a better way to distinguish them under the microscope, e.g., the right part of Figure 4(a) shows the composition of S1 after etching, whereas the left hand image shows the unetched sample.

Secondary electron images for the samples (a) S1 (SiO2 = 3.12 wt pct), (b) S2 (SiO2 = 4.01 wt pct), (c) S3 (SiO2 = 4.90 wt pct), and (d) S4 (SiO2 = 6.11 wt pct), and part of S1 was etched by hydrofluoric acid. SFC = Silico-ferrite of calcium, CF = CaO·Fe2O3, F = Fe2O3

Results of EDS for the samples (a) S1 (SiO2 = 3.12 wt pct) after etching by hydrofluoric acid, (b) S2 (SiO2 = 4.01 wt pct), (c) S3 (SiO2 = 4.90 wt pct), and (d) S4 (SiO2 = 6.11 wt pct)
Figure 5 shows the results of EDS for the samples S1 (SiO2 = 3.12 wt pct) after etching, S2 (SiO2 = 4.01 wt pct), S3 (SiO2 = 4.90 wt pct), and S4 (SiO2 = 6.11 wt pct), respectively.
Ca K-edge XANES Analysis
The Athena package in the IFEFFIT (IFEFFIT is a suite of interactive programs for XAFS analysis, combining high-quality and well-tested XAFS analysis algorithms, tools for general data manipulation, and graphical display of data.) software[15] was used to process the XANES data of all the samples. Figure 6 shows the normalized absorption spectra of Ca K-edge XANES for samples S1 to S4. The peaks a, b, and c are associated with the bound excited states and have been attributed to the electron transitions from 1s to 3d, 4p, and 5s orbitals, respectively.[16] Peak b is the position of absorption edge at 4040 eV. The absorption edge of metal Ca K-edge is at 4038 eV, which is due to that Ca exists in the samples in the +2 oxidation state.[17–20] The peak a, because of its dipole-forbidden nature of 1s to 3d transition, is quite weak and behaves as a shoulder of the peak b, and this proves that the first coordination for Ca in SFC is octahedron-ligand composed by Ca with six O.

Normalized Ca K-edge X-ray absorption spectra for S1 (SiO2 = 3.12 wt pct), S2 (SiO2 = 4.01 wt pct), S3 (SiO2 = 4.90 wt pct), and S4 (SiO2 = 6.11 wt pct) after annealed at 1473 K (1200 °C) for 24 h, respectively
The spectra intensity of S1 (SiO2 = 3.12 wt pct) and S4 (SiO2 = 6.11 wt pct) appears to change at the positions of peaks c (4046 eV) and f (4115 eV), respectively. It can be considered that the coordination environment of Ca changes in sample S1 because of the extra CaFe2O4 existence with the spectra intensity of peak c enhanced, while the appearance of Fe2O3 in S4 also illustrates that the type of the coordination atoms around Ca has changed, and the trend of the X-ray absorption oscillation at peak f changes with energy from the effect of the different atomic scattering. In addition to the peak c derived from the bound excited states, the photoelectrons with low kinetic energy (10 to 40 eV) are strongly backscattered by neighboring atoms, generating multiple scattering resonance.[21] Theoretically, such resonance is reflected in a higher energy region in an XANES spectrum, but it actually overlaps in part with the peak c, exerting minor effect on the peak b. The above result of Ca K-edge XANES analysis contains the structural information of CaFe2O4. Moreover, except for the effect of CaFe2O4, the following further analysis will conduct only on samples S2 to S4.
The change of the absorption spectra above 4050 eV (peaks d, e and f) is mainly caused by “multiple scattering contributions”[22–24] of adjacent coordination atoms. The XAS data were analyzed through the Fourier filter at k 3-weighted oscillation. Figure 7 shows the processed data. x(k) is the summation of x j(k), which represents the effect of the different coordination layers (j) on the X-ray absorption; differences in the coordination layer means that there are different ligands or different coordination distances (R). Both the position and strength of an absorption peak (shown in Figure 7) are sensitive to the change in the coordination sphere of the Ca center. The change of the position of peak e indicates that the bond length of coordination layer has increased with increase in SiO2 concentration, and the obvious change of intensity of peak f in sample S4 (SiO2 = 6.11 wt pct) indicates that the coordination environment around Ca is greatly changed by the additional SiO2 solution. However, the specific contributions of the coordination layer to the changes cannot be determined by the analysis of Figure 7 since x(k) is not only a function of photon energy (E) or wave-vector (k), but also a function of coordination distance (R).Therefore, the analysis of X-ray absorption spectra is focused on the high photon energy part of the relationship between x(k) and R by the Fourier transform (FT) in order to understand the contribution of each coordination layer on the absorption and the structural characteristics of SFC.

XAS data of the samples S2 (SiO2 = 4.01 wt pct), S3 (SiO2 = 4.90 wt pct), and S4 (SiO2 = 6.11 wt pct) processed by Fourier filter at k 3-weighted oscillation. The positions of peaks e and f are indicated by the arrows
Figure 8 shows the FT derived from k 3-weighted XANES oscillations in the k range of 2.0 ~ 12.5 Å−1 for the three samples. The FT corresponds to the radial distribution function. There are three coordination shells appeared where the peak positions are located at 0.75 to 2.25 Å, 2.25 to 3.0 Å, and 3.0 to 4.5 Å respectively. In addition, the peak widths of each shell are wide and overlap between the first and the second shells caused by the complex coordination environment, and the coordination of each shell will be analyzed using the Artemis package in IFEFFIT software combined with the crystal structure of SFC as follows.

Fourier-transformed Ca K-edge XANES (magnitude (solid lines) and imaginary part (dash lines)) of S2 (SiO2 = 4.01 wt pct), S3 (SiO2 = 4.90 wt pct), and S4 (SiO2 = 6.11 wt pct) and comparison with shell-fits (open dots, fit parameters in Table II) of S2, S3, and S4. The vertical dash lines indicate the intermediate position of each shell
Figure 9 shows the neighboring atoms around Ca in the crystal structure of SFC as characterized by the computer program Diamond 3.2i.[25] It can be seen that the complex coordination environment is constituted by Ca-O, Ca-Fe, Ca-Si, and Ca-Ca correlations. The type and number of the neighboring atoms around Ca would gradually change with the degree of Ca and Si solution. The shell-fits of these three groups of samples (S2, S3, and S4) were calculated using the Artemis package in IFEFFIT software with Ca2.9Fe10.5Si0.6O20 (The crystal structure has been determined by Ding et al.[14] ) selected as the initial structure model. The shell-fits magnitude and imaginary part are shown in Figure 8, and the results from corresponding shell-fits are provided in Table II. Normally, the two coordination layers can be distinguished in patterns after FT (shown in Figure 8) when they are 0.4 Å apart. So, the first shell is attributed to the Ca-O1st and Ca-O2nd bonds, the second shell is attributed to the Ca-O3rd and Ca-Ca1st bonds, and the third shell is attributed to the Ca-Ca2nd, Ca-Fe and Ca-Si bonds (shown in Table II).

The crystal structure of SFC and the coordination around Ca. The larger diameter red ball represents the nearest neighbor O atom, the positions of the first layer of Ca (Ca-O1st), and all Si located in the crystal structure of SFC represent the solution location. In addition, Ca-O2nd and Ca-O3rd are different and Ca-O2nd belongs to the octahedron-ligand composed by Ca with six O
The lengths of all bonds (see Table II) in each shell have grown with increasing solution of the smaller ionic radius Si4+. In contrast, the increase of the length of Ca-Ca2nd bond (here, Ca is involved in the composition of the crystal structure of SFC, see Figure 1) is large, nearly 5.0 pct, and which corresponding to the longer bond length in third shell in S4 as shown in Figure 8. The reasons for this change can be considered due to the increase of the smaller length of Si-O bond around the Ca channel (formed by the stacking of Ca-Ca2nd along c-axis, see Figure 9) with Si solution and the repulsion between Ca-Ca being enhanced. In addition, the bond lengths of Ca-O2nd and Ca-O3rd are not obviously changed by the additional SiO2 solution due to the irregular octahedron formed by Ca which in Ca channel connected with six O. It is speculated that the instability of Ca channel may be the fundamental reason for why the structure of SFC is not stable with the increase in the content of SiO2.
In addition, the shell-fit of S4 (SiO2 = 6.11 wt pct) gave coordination numbers of 5.4 and 2.2 (these numbers are associated with the site occupancy of Ca solution in SFC, and all coordination numbers are the average values calculated using the Artemis package in IFEFFIT software) for Ca-O1st and Ca-Ca1st (here, Ca represents the solution in the crystal structure of SFC, see Figure 1), respectively, which correspond to the higher first-shell and second-shell peaks as shown in Figure 8. The shell-fits of S2, S3, and S4 indicated that the coordination numbers of Ca-Fe are gradually reduced and those of Ca-Si increased, and these numbers are associated with the site occupancies of Fe (reduced) and Si (increased), respectively. They correspond to atoms nearest to the Ca channel, which indicate that the position of Si solution around the Ca channel.
These experimental data evidenced that the structural changes in SFC is due to Si solution in the center of the upper and lower tetrahedral structure near a Ca channel from our works.[14] In addition, it also proved the conclusion of Pownceby et al.[4–7] that the maximum solid solution range in SFC occurs between compositions containing approximately 7.0 to 11.7 wt pct C4S3 component along CF3-C4S3 (about 3.12 and 5.21 wt pct SiO2), and Fe2O3 began to appear when the content of SiO2 exceeded 5.21 wt pct (6.11 wt pct) in this work due to the instability of Ca channel in the crystal structure of SFC.
Conclusions
The structure of SFC synthesized in air at 1473 K (1200 °C) has been studied using a combination of XRD, FESEM, and XAS. The homogeneous SFC phase was synthesized by varying SiO2 content to investigate the effect of SiO2 on the structure of the SFC. The results are summarized as follows:
-
1.
The maximum solid solution limit of SiO2 in the crystal structure of SFC does not exceed 6.11 wt pct at 1473 K (1200 °C), and SFC formed with composition of approximately 54.05, 12.32, 2.55, and 31.08 wt pct of Fe, Ca, Si, and O, respectively, and Fe2O3 begins to appear under these conditions.
-
2.
The process of SiO2 solution is associated with the Ca channel composed of Ca octahedron in the structure of SFC, which is confirmed by the measurements of Ca K-edge X-ray absorption spectra, and Si mainly occupies the center positions of the upper and lower tetrahedra adjacent to the Ca channel. The length of Ca-Ca bond in Ca channel increases significantly with the increase of Si content. The stability of the SFC structure may be related to the changes in the instability of Ca channel.
References
T. Ikeda, K. Inoue, T. Uenaka, and M. Kanemoto: Tetsu-to-Hagané, 1981, vol. 67, pp. 726–35.
K. Inoue and T. Ikeda: Tetsu-to-Hagané, 1982, vol. 68, pp. 2190–99.
J.D.G. Hamilton, B.F. Hoskins, W.G. Mumme, W.E. Borbidge, and M.A. Montague: Neues Jahrb. Miner. Abh., 1989, vol. 161, pp. 1–26.
M.I. Pownceby, J.M.F. Clout, and M.J. Fisher-White: Trans. Inst. Min. Metall. (Sect. C), 1998, vol. 107, pp. C1–10.
M.I. Pownceby and T.R.C. Patrick: Eur. J. Mineral., 2000, vol. 12, pp. 455–68.
M.I. Pownceby and J.M.F. Clout: Trans. Inst. Min. Metall. (Sect. C), 2000, vol. 109, pp. C36–48.
T.R.C. Patrick and M.I. Pownceby: Metall. Mater. Trans. B, 2002, vol. 33B, pp. 79–89.
N.V.Y. Scarlett, M.I. Pownceby, I.C. Madsen, and A.N. Christensen: Metall. Mater. Trans. B, 2004, vol. 35B, pp. 929–36.
N.V.Y. Scarlett, I.C. Madsen, M.I. Pownceby, and A.N. Christensen: J. Appl. Crystallogr., 2004, vol. 37, pp. 362–68.
N.A.S. Webster, M.I. Pownceby, I.C. Madsen, and J.A. Kimpton: Metall. Mater. Trans. B, 2012, vol. 43B, pp. 1344–57.
N.A.S. Webster, M.I. Pownceby and I.C. Madsen: ISIJ International, 2013, vol. 53(8), pp. 1334–40.
D.H. Lister and F.P. Glasser: Brit. Ceram. Trans. J., 1967, vol. 66, pp. 293–305.
W.G. Mumme: Neues Jahrb. Miner. Abh., 2003, vol. 178, pp. 307–35.
X. Ding and X.-M. Guo: Metall. Mater. Trans. B, 2014, vol. 45B, pp. 1221–31.
B. Ravel and M. Newville: J. Synchrotron Rad., 2005, vol. 12, pp. 537–41.
L.-W. Du, S. Bian, B.-D. Gou, Y. Jiang, J. Huang, Y.-X. Gao, Y.-D. Zhao, W. Wen, T.-L. Zhang, and K. Wang: Crystal Growth & Design, 2013, vol. 13(7), pp. 3103–9.
K. Asokan, J.C. Jan, J.W. Chiou, W.F. Pong, M.-H. Tsai, Y.K. Chang, Y.Y. Chen, H.H. Hsieh, H.-J. Lin, Y.W. Yang, L.J. Lai, and I.N. Lin: Journal of Solid State Chemistry, 2004, vol. 177, pp. 2639–43.
D.R. Neuville, L. Cormier, A.-M. Flank, V. Briois, and D. Massiot: Chemical Geology, 2004, vol. 213, pp. 153–63.
O Haas, Chr Ludwig, U Bergmann, RN Singh, A Braun, T Graule (2011) Journal of Solid State Chemistry. 184, 3163-71.
I. Tanaka and T. Mizoguchi: J. Phys.: Condens. Matter, 2009, vol. 21, pp. 1–9.
A. Bianconi: XANES spectroscopy, 1988, pp. 573–662.
D. Eichert, M. Salome, M. Banu, J. Susini, and C. Rey: Spectrochim. Acta B: Atom. Spectrosc, 2005, vol. 60, pp. 850–8.
E. Paris and T.A. Tyson: Phys. Chem. Minerals, 1994, vol. 21, pp. 299–308.
B. Gilbert, B.H. Frazer, A. Belz, P.G. Conrad, K.H. Nealson, D. Haskel, J.C. Lang, G. Srajer, and G.D. Stasio: J. Phys. Chem. A, 2003, vol. 107, pp. 2839–47.
K. Brandenburg: DIAMOND, Crystal Impact GbR, Bonn, Germany, 1999.
Acknowledgments
The authors thank the National Natural Science Foundation of China for providing the financial support (Grants U146020005 and 51374017).
The XANES beam time was granted by 4B7A end-station of Beijing Synchrotron Radiation Facility, Institute of High Energy Physics Chinese Academy of Sciences. The staff members of 4B7A are acknowledged for their support in undertaking these measurements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted August 6, 2014.
Rights and permissions
About this article

Cite this article
Ding, X., Guo, XM., Ma, CY. et al. Effect of SiO2 on the Crystal Structure Stability of SFC at 1473 K (1200 °C). Metall Mater Trans B 46, 1146–1153 (2015). https://doi.org/10.1007/s11663-015-0313-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-015-0313-2