
Abstract
The solubility of vanadium oxide in the Al2O3-CaO(30 mass pct)-SiO2 system and Al2O3-CaO(35 mass pct)-SiO2 system was determined experimentally at 1873 K (1600 °C) and at a fixed oxygen potential of 9.37 × 10−11 bar. EPMA microanalyses were employed to identify the phases and their compositions in the quenched samples. The solubility of vanadium oxide in the liquid phase was found to decrease with increasing CaO content in the liquid. The vanadium oxide solubility was especially low when both CaO and Al2O3 contents were high in the liquid phase. The maximum solubility of vanadium oxide was up to 7 mass pct (as V). Two solid phases were found, a solid solution of Al2O3 and vanadium oxide and an Al2O3-rich solid phase with 16.7 mass pct V2O3. The Al2O3 solubility in the solid solution was found to increase with increasing Al2O3 content in the liquid, the impact of the CaO content in the liquid on the solubility of Al2O3 in V2O3 was found to be small. The Al2O3-rich solid phase was identified as the mineral hibonite with fractionation of V into the crystal structure.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
According to environmental legislation in some countries, e.g., Sweden, external use of steel slag from ore-based steelmaking is restricted primary due to its content of vanadium (V) and chromium (Cr). The aggregated amount of V in discharged steel slag in the Nordic countries is yearly close to 8000 tons. Earlier work has investigated the possibility to extract V into a slag phase by bubbling carbon dioxide (CO2) into a V-alloy produced by reduction of V-bearing steel slags.[1] An initial slag having the composition of 40 mass pct (percent) Al2O3, 25 pct CaO, and 35 pct SiO2 was found suitable to enhance the oxidation of V and limit the oxidation of iron (Fe) and phosphorus (P) in the oxygen partial pressure range between 1.46 × 10−11 and 1.31 × 10−10 bar.[1,2] The V-solubility in liquid in the Al2O3-CaO(25 pct)-SiO2 system was determined experimentally to be maximum up to 7 mass pct (as V) at 1873 K (1600 °C) and at a fixed oxygen potential of 9.37 × 10−11 bar.[1] Samples above the saturation point were found to be in a 2-phase equilibrium region consisting of a liquid phase and a solid solution of aluminum oxide (Al2O3) and vanadium oxide (VA phase).[1] The existence of the VA phase resulted in high viscosity of the slag.[1,2] Since the fluidity of the slag is very important for the development of the process of vanadium extraction, the phase diagram information is essential for the design of process for vanadium extraction.
The present work aims at an experimental determination of the V-solubilities in slags. It focuses on the V-solubility in Al2O3-CaO(30 pct)-SiO2 and Al2O3-CaO(35 pct)-SiO2 system at 1873 K (1600 °C) and at a fixed oxygen potential of 9.37 × 10−11 bar.
Experimental Setup and Procedure
A detailed description of the equipment and the experimental procedure can be found in a previous publication.[1] Only a salient feature of the setup is given here to orientate the readers. Al2O3, CaO, and SiO2 powders were dried at 1273 K (1000 °C) for 24 hours to remove moisture and CO2. The V2O5 powder was dried at 373 K (100 °C) for 24 hours. The suppliers and purities of the powders are listed in Table I. Samples were then prepared by mixing appropriate ratios of the dried oxide powders in an agate mortar. The ratios of the mixed powders for all samples are shown in Table II. Each sample weighed about 1 g. Either Platinum (Pt) or Molybdenum (Mo) crucibles were used to hold the samples. Pt-crucibles were made from a tube (OD: 10 mm) with thin wall by pressing the bottom together and folding the pressed part twice. The height of the Pt-crucibles was about 25 mm. Mo-crucibles with a dimension of OD: 10 mm, ID: 8 mm, and H: 25 mm were primary used, as platinum was found to dissolve in slags with high SiO2 content. The oxide powder was packed into the crucibles with the aid of a small Fe-rod. Pt-crucibles were first placed inside high density alumina crucible to prevent contact between platinum and the sample holder made of molybdenum.
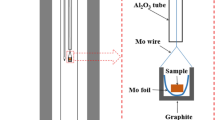
The experimental setup is presented in Figure 1. A vertical resistance furnace with MoSi2 heating elements and Al2O3 working tube (OD: 69.9 mm, ID: 63.5 mm, H: 1000 mm) was used. In order to quench the sample rapidly, the alumina reaction tube was interconnected to a water-cooled brass tube. This arrangement allowed the sample being quenched without withdrawal from the furnace. A molybdenum sample holder was connected to a molybdenum rod which was connected to a double-walled water-cooled stainless steel tube. The stainless steel tube was connected to a lifting unit. A B-type thermocouple was placed <10 mm below the sample holder for temperature measurement.

Schematic presentation of the experimental setup
The samples were then lowered down using the lifting system to the even temperature zone of the reaction chamber. The reaction chamber was sealed by the use of O-rings. The system was evacuated with a vacuum pump and thereafter filled with high-purity argon. This procedure was repeated at least 3 times. After the last evacuation, the chamber was filled with a pre-mixed CO–CO2(2.3 vol. pct) gas mixture. The oxygen partial pressure for this gas mixture was calculated to be 9.37 × 10−11 bar with an uncertainty of ±3.8 × 10−12 at 1873 K (1600 °C).[1] Throughout the heating and equilibration period, the CO–CO2 gas mixture was flushed through the chamber.
The samples were heated up at a rate of 2 K/min. After reaching 1873 K (1600 °C), the sample was kept in the furnace for 48 hours before quenching. A flow of CO–CO2 gas mixture was maintained at a flow rate of about 0.1 L/min at standard ambient temperature [298 K (25 °C)] and pressure (1 bar) (hereinafter referred to as NL/min) throughout the whole equilibrating period. Argon gas was flushed into the cooling chamber to enhance the quenching, with a flow rate of 5 NL/min. A thermocouple of type S was positioned about 10 mm above the sample holder in the quenching chamber. The thermocouples showed that the sample temperature was brought down below 1000 K (727 °C) very fast, which was far below the solidus temperature.
Electron probe microanalysis was carried out on the cross-section of each sample to gain accurate quantitative information of the phases. For this purpose, a Jeol JXA-8100 microprobe equipped with four Wavelength-Dispersive Spectrometer (WDS) was employed.
Results
The reliability of the present experimental technique is presented in a previous work.[1] Totally 11 samples were investigated for the Al2O3-CaO(30 pct)-SiO2-VO x system and totally five samples for the Al2O3-CaO(35 pct)-SiO2-VO x system. All samples were equilibrated at 1873 K (1600 °C) and at an oxygen potential of 9.37 × 10−11 bar.
Totally three phases are identified in the samples, namely (1) liquid phase, (2) a solid solution of Al2O3 and vanadium oxide named as VA, and (3) an Al2O3-rich solid phase with 16.7 mass pct V2O3 named as CAV. It is well known that the fractions of vanadium cations vary with the oxygen potential. V3+ and V4+ would be dominant at low oxygen partial pressure and low basicity in metallurgical slags. In order to present the result, V2O3 is used to describe the compositions of both liquid and solid phase.
The samples consisted either of only a liquid phase (L) or a 2-phase equilibrium of L and the solid VA phase or a 3-phase equilibrium of L, solid VA phase, and solid CAV phase.
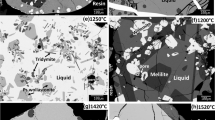
As an example, Figure 2 presents the SEM microphotograph for a sample with only liquid phase; Figure 3 presents the SEM microphotograph of the 2-phase equilibrium of L and VA; and Figure 4 presents the SEM microphotograph of the 3-phase equilibrium of L, VA, and CAV.

Microphotograph of sample no. 1 consisting of 1-phase, i.e., the liquid (L) phase

Microphotograph of sample no. 7 showing the 2-phase equilibrium of the liquid—(L) and the solid V2O3-Al2O3 phase (VA). The supercooled continuous liquid phase (L) is dark gray and the solid VA phase is light gray

Microphotograph of sample no. 3 showing the 3-phase equilibrium of the liquid (L), the solid V2O3-Al2O3 phase (VA), and the solid CaO-Al2O3-V2O3 phase (CAV). The supercooled continuous liquid phase (L) is dark gray, the solid VA phase is light gray and the solid CAV phase is black
The phases found in the Al2O3-CaO-SiO2-V2O3 system as well as the average compositions (over six measured values) of the liquid phase are presented in Table III. In the table, L stands for the liquid phase, and VA and CAV for the solid phases. V2O3 is used to describe the compositions of both liquid and solid phase. Note that the compositions are normalized values. Normalization was done by taking the sum of the four components as 100 pct.
The relative average deviation (RAD) of the EPMA analysis for compounds with more than 2 pct is also presented in Table III. RAD is defined by Eq. [1],
where x i , …, x N represent the data points, \( \overline{x} \) is the average value of the data, and N is the number of the total data points. The relative average deviation for the data points was generally <2 pct for Al2O3, CaO, and SiO2 and <6 pct for V2O3 in the liquid. The composition of the solid phases are listed in Table IV.
Discussion
Experimentally Determined Liquidus Surface
The compositions of the samples were aimed at 30 and 35 pct CaO, respectively. However, for various experimental reasons, the CaO contents deviated somewhat from these targeted values. While the exact experimentally determined compositions are listed in Table III, to present the liquidus surface in a graphical manner, normalization is needed. The normalization was carried out according to the following procedure; viz. the CaO content within a range of 27.5 to 32.5 pct CaO was adjusted to 30 pct, while the concentrations of the remaining components were normalized in proportion to their original fractions with their sum being 70 pct. The CaO content within a range of 32.5 to 37.5 pct CaO was adjusted to 35 pct and the sum of the remaining components to 65 pct. Note that the readers should use the data listed in Table III for any thermodynamic calculation.
Figures 5 and 6 present the experimentally determined liquidus surface at 1873 K (1600 °C) in the section of 30 and 35 pct CaO, respectively. The liquidus surface in the section of 25 pct CaO, determined experimentally in an earlier study,[1] is reproduced in Figure 7. Note that these relationships are valid at an oxygen potential of 9.37 × 10−11 bar. In these figures, the liquid phase is marked with the symbol “●”. The coexistence of the two phases L and VA is marked with the symbol “▲”. The coexistence of the three phases L, VA, and CAV is marked with the symbol “■”. The line represents the phase boundary of the liquid–solid mixture, i.e., slags with a composition along the line are saturated with V, while V is completely dissolved in slags with compositions below this line. Along each side of the triangles the three component systems are given by the 30 and 35 pct CaO sections of the Al2O3-CaO-SiO2, Al2O3-CaO-V2O3, and CaO-SiO2-V2O3 system. However, only data for the Al2O3-CaO-SiO2 system can be found in the literature.[3] The liquidus surface for the 30 pct section in the Al2O3-CaO-SiO2 system at 1873 K (1600 °C) is marked with the symbol “□”.

Graphical presentation of the liquidus composition projected on the section of 30 pct CaO in the quaternary Al2O3-CaO-SiO2-V2O3 system at 9.37 × 10−11 bar and 1873 K (1600 °C). The liquid phase is marked with the symbol “●”. The coexistence of the two phases L and VA is marked with the symbol “▲”.The coexistence of the three phases L, VA, and CAV is marked with the symbol “■”. The liquidus surface for the 30 pct CaO section in the Al2O3-CaO-SiO2 system at 1873 K (1600 °C) is marked with the symbol “□”

Graphical presentation of the liquidus composition projected on the section of 35 pct CaO in the quaternary Al2O3-CaO-SiO2-V2O3 system at 9.37 × 10−11 bar and 1873 K (1600 °C). The liquid phase is marked with the symbol “●”. The coexistence of the two phases L and VA is marked with the symbol “▲”

Graphical presentation of the liquidus composition projected on the section of 25 pct CaO in the quaternary Al2O3-CaO-SiO2-V2O3 system at 9.37 × 10−11 bar and 1873 K (1600 °C). The liquid phase is marked with the symbol “●”. The coexistence of the two phases L and VA is marked with the symbol “▲”. The liquidus surface for the 25 pct CaO section in the Al2O3-CaO-SiO2 system at 1873 K (1600 °C) is marked with the symbol “□”
The solubility of V in the liquid phase decreases with increasing CaO content in the liquid. The V-solubility is especially low at both high CaO and Al2O3 content in the liquid phase.
Precipitation of Solid VA Phase
In a previous work investigating the Al2O3-CaO(25 pct)-SiO2 system,[1] the Al2O3 solubility in V2O3 was found to increase with increasing Al2O3 content in the liquid as the Al2O3 activity in the liquid increases with the increase of Al2O3 in the liquid phase. In Figure 8, it is shown that the same correlation is also valid for the Al2O3-CaO(30 pct)-SiO2 and Al2O3-CaO(35 pct)-SiO2 system, viz. the Al2O3 solubility in V2O3 increases with increasing Al2O3 content in the liquid. In the figure, samples are grouped with respect to the CaO content in the liquid. The impact of the CaO content in the liquid on the solubility of Al2O3 in the V2O3 phase is small, especially when the Al2O3 content in the liquid is <30 pct. Note that it is difficult to investigate the correlation above 30 pct Al2O3 in the liquid at higher CaO contents as the V-solubility in the liquid is very low.

Al2O3 in V2O3 (VA phase) as a function of the Al2O3 content in the liquid. Samples are grouped with respect to the CaO content in the liquid
Precipitation of Solid CAV Phase
The 3-phase equilibrium of L, solid VA phase, and a solid CAV phase was found in the Al2O3-CaO(30 pct)-SiO2 system (sample no. 3) at high Al2O3 content in the liquid. The components in the CAV phase were Al2O3 (74.6 pct), CaO (8.2 pct), SiO2 (0.5 pct), and V2O3 (16.7 pct). In order to confirm the finding, this compound was also studied separately by synthesis. The starting materials used to prepare this compound have earlier been presented in Table I. The preparative technique involved careful grinding of equivalent quantities of the component oxides in an agate mortar and sintering pressed pellets. Two pellets with identical weight in composition were sintered in Pt-crucibles using the same experimental setup and procedure earlier described, i.e., sintering at 1873 K (1600 °C) and at oxygen partial pressure of 9.37 × 10−11 bar for a duration of 48 hours. A sample of each pellets were mounted into conductive resin with carbon deposited in vacuum on the surface and analyzed with SEM–EDS analysis (Zeiss Merlin FE-SEM with an Oxford EDS detector) at 20 kV and 1 nA. The remaining parts of the two pellets were ground using an agate mortar and then used for determination of the crystallographic parameters using a Panalytical Empyrian Diffractometer, equipped with a Pixcel 3D detector and Cu-Kα radiation. The XRD pattern was recorded from 5 to 90 deg sin2θ at 0.026 deg/step and 88 s/step.
The results of the SEM–EDS analysis for the two pellet samples as well as the composition of the solid CAV phase found in sample no. 3 using WDS analysis are presented in Table V. The WDS analysis of sample no. 3 reveals a stoichiometric relation that is identical to calcium hexaluminate (CaAl12O19 or CaO·6Al2O3) which occurs in nature as the mineral hibonite. This compound is also reported to crystallize from liquid Al2O3-CaO-SiO2 melts with high Al2O3 contents.[3]
The mineral hibonite has magnetoplumbite-type structure with the general crystallochemical formula of A(12)M1(6)M2 (5)2 M3 (4)2 M4 (6)2 M5 (6)6 O19. Calcium occurs in 12-fold coordination (site A), whereas Al3+ ions are distributed over three distinct octahedral sites (M1, M4, and M5), one tetrahedral site (M3) and a trigonal bipyramidal site providing five-fold coordination by oxygen ions (M2). Hibonite has also the ability to accommodate a wide variety of ions, with different valence and coordination including V.[4] Despite this ability insignificant deviation from the stoichiometry: Ca = 1; (Al + V + Fe + Cr + Mg + Ti + Si + Sc) = 12 and O = 19 is usually reported.[5] Only a few references indicate that some meteoritic hibonites are highly nonstoichiometric.[6,7]
The diffraction patterns of the two pellet samples are shown in Figure 9. As shown in the figure, the crystal is identified as the mineral hibonite 5H with the chemical formula CaO((AlFe)O3)6.[8] It can, therefore, be concluded that the CAV phase found in this study is in fact fractionation of V into hibonite.

Diffraction pattern of synthesized CAV phase
One reason that the CAV phase was not found in sample 10, situated close to sample no. 3 in Figure 6, could be due to that the ratios of the mixed powders for the two samples are different. The weighed in composition of Al2O3 in sample no. 3 (3-phase equilibrium of L, VA, and CAV) was 44.24 pct and in sample no. 10 (2-phase equilibrium of L and VA) 33.29 pct while at the same time the Al2O3 content in the liquid was quite similar, 36.6 and 35.4 pct, respectively. This surplus of Al2O3, except for the Al2O3 going into solid solution (VA phase), could be the driving force for forming the Al2O3-rich CAV phase.
Conclusions
The V-solubility in the Al2O3-CaO(30 mass pct)-SiO2 and Al2O3-CaO(35 mass pct)-SiO2 system at 1873 ± 4 K (1600 °C) and at a fixed oxygen potential (9.37 × 10−11 ± 3.8 × 10−12 bar) was determined experimentally. EPMA analyses were made to identify the phases present and their compositions. The main findings could be summarized as:
-
1.
The solubility of V-oxide increases with decreasing Al2O3 and CaO content in the liquid.
-
2.
The maximum V-solubility is up to 7 mass pct (given as V).
-
3.
Samples saturated with V-oxide are either in a 2-phase region consisting of liquid (L) and a solid phase (VA), or in a 3-phase equilibrium of L, solid VA phase, and a solid Al2O3-rich phase (CAV).
-
4.
The VA phase is a solid solution of Al2O3 in V-oxide. The Al2O3 solubility in this solid solution was found to increase with increasing Al2O3 content in the liquid. As maximum, about 30 mass pct Al2O3 was detected. The impact of the CaO content in the liquid on the solubility of Al2O3 in V2O3 was found to be small.
-
5.
The CAV phase was identified as the mineral hibonite with fractionation of V (16.7 mass pct as V2O3) into the crystal structure.
References
M. Lindvall, J. Gran, and D. Sichen: CALPHAD 2014, vol. 47, pp. 50–55.
M. Lindvall, E. Rutqvist, G. Ye, J. Bjorkvall, and D. Sichen, Steel Res. Int., 2010, vol. 81, pp. 105–11.
E.F. Osborn and A. Muan: Phase Equilibrium Diagrams of Oxide Systems, American Ceramic Society, Ohio, 1960, Plate 1.
R. Burns and V. Burns: J. Geophys. Res., 1984, vol. 89, pp. C313–21.
J. Armstrong, G. Meeker, J. Huneke, and G. Wasserburg: Geochim. Cosmochim. Acta, 1982, vol. 46, pp. 575–95.
G. Macpherson, M.M. Bar-Matthews, T. Tanaka, E. Olsen, and L. Grossman: Geochim. Cosmochim. Acta, 1983, vol. 47, pp. 823–39.
A. Goresy and P. Ramdohr: Meteoritics, 1980, vol. 15, p. 286.
M. Harder and H. Mueller-Buschbaum: Z. Naturforsch. B Anorg. Chem. Org. Chem., 1977, vol. 32, pp. 833–34.
Acknowledgments
Funding and support from MISTRA, SSAB, LKAB, Ruukki, SSAB Merox, and Jernkontoret are gratefully acknowledged with special thanks to Dr. G. Ye and Dr. J. Björkvall of Swerea MEFOS. Thanks also to Stefan Andersson for technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted October 16, 2014.
Rights and permissions
About this article

Cite this article
Lindvall, M., Sichen, D. Determination of Vanadium Solubility in the Al2O3-CaO(30 Mass Pct)-SiO2 and Al2O3-CaO(35 Mass Pct)-SiO2 System. Metall Mater Trans B 46, 733–740 (2015). https://doi.org/10.1007/s11663-014-0237-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-014-0237-2