
Abstract
The reduction of iron oxide fines to wustite between 590 °C and 1000 °C with a CO–CO2 gas mixture of low reducing potential was studied. The reduction kinetics and the dominating reaction mechanism varied with the temperature, extent of reduction, and type of iron oxide. Reduction from hematite to wustite proceeded in two consecutive reaction steps with magnetite as an intermediate oxide. The first reduction step (hematite to magnetite) was fast and controlled by external gas mass transfer independently of the oxide type and the temperature employed. The second reduction step (magnetite to wustite) was the overall reaction-controlling step, and the reduction mechanism varied with the temperature and the oxide type. Moderately porous oxide fines followed the uniform internal reaction for the temperature range studied. For highly porous oxides, the second reduction step was controlled by external gas mass transfer above 700 °C. Below that temperature, a mixed regime that involves external gas mass transfer and limited mixed control, which comprises pore diffusion and chemical reaction, takes place. The rate equations for this mixed control reaction mechanism were developed, and the limited mixed control rate constant (klm) was computed. For denser oxides under uniform internal reaction, the product of the rate constant and pore surface area (k·S) was calculated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Extensive research efforts have been devoted to the reduction of iron oxides for conditions pertinent to blast furnace and direct reduction processes. Considering the gaseous reduction of porous hematite to iron, attention has been focused mainly on the later stages of the reduction process, which involve reactions to metallic iron as this is the overall slowest reduction step.[1,2] In these conditions, gases of high reducing potential are employed such that the early stages of reduction (from Fe2O3 to FeO) are fast and often influenced by mass transport mechanisms.
Limited information is available for the initial stages of the reduction process that involve iron oxide fines under highly oxidized atmospheres and moderate temperatures in which Fe2O3 or Fe3O4 can be reduced to FeO but not to metallic iron. These conditions are relevant to bath smelting processes using coal directly to produce hot metal such as HIsmelt, DIOS, and AISI in which the process off-gas (primarily CO–CO2–H2–H2O) may be used for prereduction of iron ore. They also are relevant to a newly proposed smelting process in which coal-char is employed, and an off-gas consisting mainly of CO-CO2 is generated.[3] The coal-char is obtained in a separate process in which the hydrogen from coal is captured. The CO–CO2 smelter off-gas is used conceptually to preheat and partially reduce iron oxide fines in a prereduction unit. The smelter off-gas composition or the off-gas reducing potential is determined by the oxygen source (O2, or air) and the extent of conversion of CO to CO2 in the smelter, i.e., the post combustion degree (PCD), defined by Eq. [1] as follows:
Post combustion is an important smelter parameter because it has the potential to reduce coal consumption in the process.[4] Generally, higher PCDs may be achieved for smelters operating with coal-char compared with coal.[3,5] An ideal situation is a PCD of about 50 pct, which allows for an adequate supply of energy to the smelter while preserving limited reducing power in the off-gas to convert Fe2O3 to FeO.[4] This reduction degree corresponds to the removal of about one third of the oxygen in Fe2O3 and addresses a use of the chemical and sensible energy available in the smelter off-gas.
The objective of this study was to investigate the kinetics and mechanisms of reduction of porous iron oxide fines to FeO at moderate temperatures with a simulated smelter off-gas of low reducing potential. In this study, the reaction gas is limited to an equimolar CO–CO2 mixture, which would be the case of a coal-char smelter operating at 50 pct PCD. If coal—which typically contains 4 to 5 wt pct hydrogen[6]—is used in the smelter, then the off-gas could contain up to about 30 vol pct of H2 and H2O. This was not considered in the present study.
Background and Reduction Models
The gaseous reduction of iron oxides has been the subject of extensive research, and a thorough review would extend beyond the possibilities of a single text. Therefore, only the principal aspects of the reduction mechanisms pertinent to the present work are covered briefly. The reduction reactions of interest are represented by Reactions 2 and 3 for Fe2O3 and Fe3O4, respectively.
The overall reduction involves the conversion of Fe2O3 to FeO through the intermediate Fe3O4. The Fe2O3 → Fe3O4 → FeO reaction sequence may proceed in a well-defined stepwise mode in which the Fe2O3 → Fe3O4 reduction step essentially is completed before the Fe3O4 → FeO takes place or in a simultaneous consecutive mode, in which Fe2O3, Fe3O4, and FeO coexist for significant periods of time. The earlier usually is favored by a moderate reaction driving force and by porous oxide particles,[7] which are characteristics of the present study.
In porous oxides, most of the area available for a reaction exists internally within the pore structure of the particles. The pore structure and, consequently, the internal surface area influences the rate and mechanism of reduction.[8] Several mechanisms may control the overall rate of gaseous reduction of porous iron oxides, and detailed reviews have been published by Turkdogan and others.[1,7,9–15] Considering the reactions of interest and porous iron oxides in isothermal reaction conditions, possible rate controlling steps include the following:
-
(a)
External mass transfer of CO or CO2 from/to the bulk gas stream to/from the oxide surface through a gas boundary layer;
-
(b)
Diffusion of CO or CO2 through the oxide pore structure;
-
(c)
Heterogeneous chemical reaction at the oxide pore surface.
The overall reduction rate is determined by the slowest individual step; however, the reaction rate is frequently in a mixed control regime in which the rate is controlled simultaneously by two or more mechanisms. Heat transfer, which can also play a role in reduction processes, is not expected to influence the reduction rate for isothermal conditions, modest reaction enthalpies, or small oxide particles, which are conditions of the present work.
External Gas Mass Transfer
At high reduction temperatures, the chemical reaction rate is generally fast, and the transport of gases through a boundary layer external to the oxide surface becomes the limiting rate mechanism. A concentration gradient of gas is established outside the particle, and the removal rate of oxygen from the oxide dn O /dt is represented by Equation 4 for counter-current equimolar diffusion:
where n O and t represent the quantity of reducible oxygen in the oxide (g·atom-O) at time t (seconds); m is the mass transfer coefficient of CO–CO2 (cm/sec); A δ corresponds to the boundary layer area external to the particle (cm2); R and T stand for the gas constant (cm3·atm/mole·K) and temperature (K); \( p_{\text{CO}}^{B} \) and \( p_{\text{CO}}^{S} \) are the partial pressure of CO at the bulk stream and external particle surface, respectively (atm).
Chemical Reaction Control
This mechanism is likely for low temperatures and small porous oxide particles in which the chemical reaction resistance is much greater than the boundary layer and pore diffusion processes. Therefore, complete gas diffusion within the pores of the particles with reduction is distributed uniformly throughout the oxide so that no macroscopic receding interface is formed.[1,13] This mechanism often is referred to as the uniform internal reaction. In this regime, the reduction rate becomes proportional to the quantity of reducible oxygen in the sample (n O ) as represented by Eq. [5], which can be integrated to yield Eq. [6] as follows[16]:
The fraction of oxide reduced is represented by F, while k, S, and Keq are the intrinsic reaction rate constant (cm/sec), pore surface area per unit mass of oxygen (cm2/g·atom-O), and equilibrium constant, respectively.
Limited Mixed Control
This regime has been observed for both carbon oxidation and iron oxide reduction[1,17] for conditions in which the chemical reaction rate becomes comparable with the rate of diffusion of gas through the pore structure. Therefore, reduction is limited by the availability of gas at a given distance from the oxide particle center. Initially, only the outer surface of the particle is in contact with the reducing gas. A reaction product layer that surrounds a core of unreacted oxide is formed, which establishes a macroscopic receding interface. In addition, because reaction and diffusion rates are similar, the receding interface has a diffuse character as reduction proceeds in the pore mouths ahead of the nominal moving interface.[1]
The reduction rate is controlled jointly by chemical reactions as well as gas diffusion through the oxide pores and is expressed in Eq. [7], which incorporates the reversible reaction term as follows[16]:
where A i represents the reacting area associated with the receding interface; \( p_{\text{CO}}^{S} \) and \( p_{\text{CO}}^{E} \) denote the surface and equilibrium partial pressures, respectively; ρ o is the reducible oxygen density (g·atom-O/cm3); and D e stands for the effective diffusivity of CO–CO2 in the pores ahead of the interface. Frequently, the limited mixed control rate constant is given by klm = (ρ o D e kS)½, with cm/sec units. In deriving the limited mixed control equation, it is assumed that gas transport through the product layer is fast so that the partial pressure at the reacting interface is equivalent to the pressure at the external surface (\( p_{{i}}^{{S}} \)) during the course of reaction. In addition, gas transfer is solely due to concentration gradients, temperature, and pressure within the oxide particles are uniform.
When considering a broad temperature range, it is necessary to account for the effect of external gas transfer on the limited mixed control rate, which is achieved through Eqs. [4] and [7]. These computations lead to Eq. [8], which considers external gas mass transfer and limited mixed control, which comprise gas transport, external and through the oxide pores, as well as heterogeneous chemical reaction:
It is important to note that the area terms in Eq. [8] (A δ and A i ) are different. A δ is associated with the external boundary layer, which is constant under certain conditions, whereas A i is related to the reacting interface of the porous oxide particle, which changes as the reduction proceeds for spherical particles. Representing n O and A i for a number (n p ) of spherical oxide particles of radius (r) and taking A δ as a constant, Eq. [9] is obtained, which is derived and rearranged yielding Eq. [10]:
Integrating, rearranging, and considering the fractional reduction (F) in Eq. [10] with respect to r and t, leads to Eq. [11], where r 0 and n O represent the initial radius of an individual oxide particle and the initial mass of reducible oxygen, respectively. Equation [11] is expressed as follows:
The second term on the right-hand side of Eq. [11] reflects the correction for external gas mass transfer on the limited mixed control. When the mass transfer coefficient (m) is much larger than the limited mixed control constant (klm), the term becomes small and Eq. [11] reduces to the usual form of the limited mixed control that is applied to a spherical geometry. The parameter determined from Eq. [11] is the limited mixed control rate constant (klm), which contains the effective diffusivity D e , specific surface area S, and intrinsic rate constant k besides the oxygen density ρ o . Thus, to obtain k or the product of the k and S—as usually is expressed for porous materials—it is necessary to evaluate the effective gas diffusivity in the oxide pores.
Effective Gas Diffusivity (De)
Because of the additional resistance to gas flow imposed by the oxide pore structure, the effective diffusivity in the pores is smaller than the molecular gas diffusivity.[11] Thus, the effect of the pore network has to be taken into account when expressing D e , and correlations have been proposed.[18–20] Depending on the relationship between pore dimension and mean free path of the diffusing gas, D e is defined by a combination of mechanisms that involve collisions between gas molecules (molecular diffusion) and gas-pore walls (Knudsen diffusion).
Previous Studies on Reduction of Hematite to Wustite
The reduction of iron oxides has been the subject of extensive research. In the reduction of Fe2O3 to metallic iron, it generally is agreed that the early stages of the reaction Fe2O3 → FeO proceed relatively fast and that the FeO → Fe reaction is the overall controlling step, so it has received considerably more attention. Rate constants for the initial stages of reduction (from Fe2O3 to FeO), therefore, are much less well established than those constants for FeO or Fe3O4 to iron. In this section, only a limited number of studies that deal with the gaseous reduction of Fe2O3 to FeO are reviewed.
Trushenski[10] studied the nontopochemical reduction of porous pellets between 600 °C and 900 °C in which the Fe2O3 → Fe3O4, Fe3O4 → FeO, and FeO → Fe reactions were studied independently and consecutively by controlling the CO–CO2 composition. Reaction rate constants were obtained by fitting the experimental results with the developed model. Activation energies of 69 kJ/mole and 78 kJ/mole were obtained for the consecutive Fe2O3 → Fe3O4 and Fe3O4 → FeO reactions, respectively. However, because of the low reacting gas flow rates employed to assure stepwise reduction, significant external mass-transfer effects were introduced on the overall reduction rate. Murayama,[21] applied the unreacted core model to hematite pellets reduced stepwisely and found activation energies of 79 kJ/mole and 121 kJ/mole for the Fe2O3 → Fe3O4 and Fe3O4 → FeO reactions, respectively.
Doherty[22] studied the isothermal reduction of hematite fines to wustite by H2–H2O, CO–CO2, and H2–H2O–CO–CO2 mixtures in a fluidized bed where mass transfer resistance is minimized. Reduction rates were reported to increase with temperature, reducing potential and H2 content in the gas, and gas flow rate. Product layer formation was observed for the Fe2O3 → Fe3O4 step, whereas uniform internal reaction was reported for Fe3O4 → FeO. Experiments were adjusted to simulate industrial practices in which fluid beds are operated close to reacting gas starvation—a condition that can influence the rate of a reaction, especially for reactions with small or moderate equilibrium constants.[23] In the early stages of reduction by H2–H2O, the fluid bed exit gas composition was close to the composition for equilibrium with Fe3O4–FeO, which may explain the dependency of the reduction rate on gas supply or flow rate. The gas utilization decreased as the reduction proceeded in CO–CO2 in which an approximate rate constant of 1.3 × 10−5 mol/atm × m2 × s was obtained for the later stages of the Fe3O4 → FeO reduction at 700 °C.
The kinetics of the Fe2O3 → FeO reduction were studied recently by Piotrowski et al.[24] from 700 °C to 900 °C, who employed Fe2O3 powder of similar size, density, and specific surface area as an oxide used in this study. The reacting gas was a mixture of CO–H2 with reducing potential to metallic iron, and the Fe2O3 → FeO step was studied using the theoretical weight loss considering the overall transformation. In general, the Fe2O3 → FeO reduction data was sigmoid-shaped and exhibited incubation, acceleration, as well as decay regions. Piotrowski et al.[24] applied the Avrami–Erofe’ev model, which assumed the random nucleation of product phase, negligible mass transfer resistances, as wel as a parabolic diffusion model. The first half of the Fe2O3 → FeO reduction data followed the Avrami model relatively well. After that, between 0.5 < F < 1, the reaction data was compatible with the one-dimensional diffusion model. Using the Avrami model strictly, an activation energy of about 58 kJ/mol was obtained; however, the contribution of H2 and CO to the reduction was unclear. Furthermore, the shift in the controlling mechanism suggested by the authors also could be related to a change in the actual reaction that took place.
Experimental
The following three types of porous iron oxide fines were used: (1) reagent grade porous analytical hematite (PAH) obtained from Fischer Chemicals (Pittsburgh, PA), (2) commercial grade Brazilian concentrate supplied by VALE (formerly CVRD, Brazil), and (3) North American Taconite provided by US Steel Technology Center (Pittsburgh, PA). Table I shows the chemical composition of the oxides as well as the specific surface area, particle densities, size ranges, and calculated porosities. The PAH particles were mostly spherical, wheras VALE and Taconite had irregular geometries. The as-received oxide samples were dried in a muffle furnace at 150 °C for about 72 hours and subsequently sieved to specific particle sizes. The sieved iron oxide was weighed and evenly spread across a cylindrical alumina crucible that ranged from 10 mm to 25 mm diameter and a fixed height of 5 mm. Sample weights from 50 mg to 400 mg of the dried and sieved fines have been used, which ensured that the oxides were arranged in only a few layers to minimize gas resistance caused by particle agglomeration.
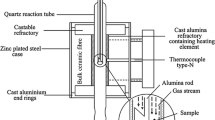
The crucible with the oxide was suspended from one arm of a microbalance (CAHN-1000, Cahn Div., Ventron Corp., Cerritos, CA) within the hot zone of a quartz reaction tube arranged in a resistance furnace. The furnace was heated to a desired reaction temperature (590 °C–1000 °C) under an inert gas atmosphere. Once a stable temperature was obtained, the inert gas was switched to the reacting CO–CO2 gas and the reduction proceeded isothermally. The CO–CO2 gas mixture had an equal volume ratio that represented a 50 pct post-combusted smelter off-gas. Aiming to minimize external mass transfer resistance, gas flow rates up to 7 Lpm were employed. Considering the gas flow rate and the experimental geometry and temperatures of the present work, gas flow rates of about 32 cm/sec could be performed. Although this rate is high for laboratory scale experiments, it is substantially lower than industrial processes, which range from about 2 to 5 m/sec.[25] Nevertheless, the gas flow rates are well above the rates necessary to avoid gas starvation because less than 1 pct of the reacting gas is used during reduction. In addition, no fluidization of the oxide powder was observed, and cooling effects by the gas were found negligible.
The weight change associated with the iron oxide reduction by the CO–CO2 gas was acquired at a scan rate of 1 Hz with an Omega data acquisition system. The raw experimental weight loss data were converted to fractional reduction according to Eq. [12], where \( n_{{O}}^{{i}} \) and \( n_{{O}}^{{t}} \) represent the reducible mass of oxygen initially and at time t, respectively. In this way, F assumes a value of “1” when the oxide is reduced fully to FeO.
Morphological examination of partially reduced oxides was conducted with a scanning electron microscope (Philips FEM-XL30; Philips Electronics, Andover, MA). Samples also were analyzed by X-ray diffraction (Rigaku Geigerflex Θ/Θ Difractometer; Rigaku, The Woodlands, TX).
Results and Discussion
Typical Experimental Results and the Stepwise Reduction
Figures 1(a) to (c) show typical results of reduction at several temperatures obtained for PAH, VALE, and Taconite, respectively. The fractional reduction for Taconite corresponds solely to the Fe3O4 → FeO reaction, whereas for PAH and VALE, the Fe2O3 → Fe3O4 → FeO reaction process is accounted for.

Fraction of oxide reduced to FeO as a time function for several temperatures at total CO/CO2 flow of 5 lpm and ∅crucible = 25 mm. (a) PAH, (b) VALE, and (c) Taconite
The kinetics of reduction is strongly dependent on the experimental temperature and the type of iron oxide that the time for complete reduction to FeO ranges from about 1 to more than 360 minutes. Also, for given reaction temperature, PAH reduces to FeO much faster than both VALE and Taconite. In addition, although thermodynamically feasible in the present experimental conditions, carbon deposition has not been observed in any of the experiments, which may be related to the absence of metallic iron surfaces (as the reduction proceeds to wustite only) as well as the lack of hydrogen in the reducing gas mixture.[26]
Two regions with distinct reaction rates are observed for PAH and VALE where an inflection seems to take place at about one third of the reduction process. In general, substantially faster reaction rates are observed before the inflection point. The decrease in reaction rate after the inflection point could be related to a change in the dominating reaction mechanism or to a shift in the actual chemical reaction that took place. To better understand this behavior, a series of experiments was carried out in which the reacting oxides were interrupted before and after the inflection point. X-ray analysis of the partially reacted oxides revealed Fe2O3/Fe3O4 and Fe3O4/FeO peaks before and after the inflection point, respectively, which indicates that the Fe2O3 → Fe3O4 → FeO process followed a sequential two-step reaction mechanism. As a result, the experimental data for PAH and VALE can be assessed separately for each individual step—Fe2O3 → Fe3O4 followed by Fe3O4 → FeO. Similar stepwise reaction mode from Fe2O3 to FeO was reported by Wagner et al.[27] in a recent reduction work of iron oxide fines to metallic iron in hydrogen. For Taconite, only the Fe3O4 → FeO reduction step took place.
Reduction of Fe2O3 to Fe3O4
Considering the overall reduction of PAH and VALE in Figures 1(a) and (b), the Fe2O3 → Fe3O4 reaction step is faster than the Fe3O4 → FeO. Furthermore, the Fe2O3 → Fe3O4 rate was controlled by external gas mass transfer under the current experimental conditions. Evidence is shown in Figure 2 where the Fe2O3 → Fe3O4 reduction rate increases with gas velocity and is nearly independent of reaction temperature. Therefore, attention is focused mainly on the Fe3O4 → FeO reaction because it is the slowest and the controlling step of the overall reduction process to wustite. The forthcoming results and discussions are limited to the Fe3O4 → FeO reaction.

Influence of reducing gas velocity calculated at reaction temperature on the rate of reduction of PAH from Fe2O3 → Fe3O4 at 620 °C, 700 °C, and 900 °C. Woxide = 100 mg and Øcrucible = 25 mm
Reduction of Fe3O4 to FeO
The effect of gas velocity on the Fe3O4 → FeO reaction rate also was studied for PAH, VALE, and Taconite. It was observed that increasing the gas flow rates only influenced the kinetics of PAH for temperatures greater than 700 °C. The effect of gas velocity on the PAH reduction rate for temperatures lower than 700 °C and for VALE and Taconite, over the temperature range of this study, was essentially negligible, however, according to Warner,[28] this indication alone cannot rule out external gas transfer control.
Therefore, the effect of external gas mass transfer on the Fe3O4 → FeO reduction step was further evaluated by comparing the observed initial reaction rates of PAH, VALE, and Taconite with those calculated with Eq. [4]. Results are shown in Figure 3 in terms of fractional reduction rates. Considering the initial Fe3O4 → FeO reaction rates, the results represent the maximum effect of external gas mass transfer on the reduction rate. In calculating the rates, A δ was taken as the crucible area, \( p_{\text{CO}}^{{B}} \) was fixed at 0.5 atm, and \( p_{\text{CO}}^{{S}} \) was approximated by the equilibrium partial pressure with Fe3O4/FeO and was computed with thermodynamic data available on FactSageTM. The mass transfer coefficient (m) was estimated by measuring the Mg vaporization under identical flow and geometrical conditions as the reduction experiments. Details of the m experimental determination are discussed in the Appendix. Furthermore, the assumption of A δ as the crucible area was substantiated by vaporization and reduction experiments under external gas mass transfer control in which the rate of reaction was proportional to the crucible area provided that a shallow bed of porous oxide was maintained.

Observed fractional reduction rates for Fe3O4 → FeO compared with calculated rates that consider external gas mass transfer for PAH, VALE, and Taconite as a function of temperature
The initial observed rates for Taconite and VALE are much lower than the calculated values within the temperature range studied. In fact, the differences between the calculated and observed rates are generally greater than an order of magnitude with the exception of VALE between 950 °C and 1000 °C, which is about six to seven times greater. This trend suggests that the mechanism that controls the Fe3O4 → FeO reduction for Taconite and VALE is not affected significantly by the transfer of gas between the bulk stream and the particle surface. A distinct situation prevails for PAH in which the difference between the calculated and experimental rates varies considerably with the temperature. For temperatures greater than 800 °C, the ratio between the calculated and observed rates is only about 1.3, whereas for temperature less than 620 °C the ratio is greater or about 10, which indicates that external gas mass transfer effects for PAH becomes progressively less dominant as reduction temperature is decreased, and seems to be negligibly small below 620 °C. Additional experiments carried with the three iron oxides in which the reacting CO–CO2 gas was diluted with Helium or Argon support these observations.[29]
Gas dilution experiments carried out at 620 °C for PAH are shown in Figure 4. The Fe3O4 → FeO reaction rate was faster in CO–CO2–He than for CO–CO2–Ar, which also was observed at lower reaction temperatures. Because external gas mass transfer resistance was negligibly small at and below 620 °C, it suggests that another mechanism dependent on gas diffusivity is controlling the Fe3O4 → FeO reduction for PAH. A possible mechanism that took place place, considering the highly porous and small oxide particles, is limited mixed control in which the reaction rate is limited jointly by gas diffusion through the oxide pores and by a chemical reaction. To verify this hypothesis, experiments were carried out in which the reduction reaction was interrupted within the Fe3O4 → FeO region at several temperatures. The internal structure of the partially reduced oxide was analyzed with a SEM. The cross sections obtained for PAH partially reduced at 620 °C are shown in Figures 5(a) and (b). For comparison, the cross sections of partially reduced VALE obtained at 700 °C also are included as Figures 5(c) and (d). All partially reduced oxides samples were analyzed by X-ray before the microscopic observations, which confirmed the exclusive presence of Fe3O4 and FeO.

Effect of inert gas dilution on the Fe3O4 → FeO reduction kinetics for PAH at 620 °C

Cross-sectional view of partially reduced PAH (a) and (b) as well as VALE (c) and (d) within the Fe3O4 → FeO reduction region
A macroscopic receding interface is evident in Figures 5(a) and (b) for PAH at 620 °C. This feature also was observed for PAH samples that were partially reduced from 590 °C to 650 °C. Although the presence of a receding interface supports the limited mixed control, other mechanisms also are associated with the occurrence of a macroscopic interface, which is the case of reactions controlled by gas diffusion through the pores of the outer product layer. However, this mechanism is not expected to predominate for PAH because the average particle size is 150 μm. In fact, an average effective diffusivity 104 times smaller than the molecular diffusivity was computed from PAH reduction data between 590 °C and 650 °C. The effective gas diffusivity for the later reduction stages of PAH was calculated with Eq. [13].
The receding interface, therefore, likely is correlated with the PAH reaction under limited mixed control in the pores. In addition, the interface seems to be “sharp,” as depicted in Figure 5(b). This characteristic probably is associated with the large surface area of PAH as the diffuse aspect of the limited mixed control interface is proportional to the relative ratio between the rates of gas diffusion and chemical reaction in the pores. Therefore, for highly porous oxides under limited mixed control regime, the surface area available for a reaction is large, which results in fast chemical reaction rates and an apparent “sharp” receding interface. Additional experiments in which PAH was sintered before reduction indicated that the extent of reduction ahead of the interface increases with sintering, which will be discussed in more detail. Considering VALE, the micrographs on Figures 5(c) and (d) show a homogeneous appearance with no macroscopic interface, which indicates that gas diffusion through the oxide pores is fast, and reduction may occur uniformly throughout the particle.
The effect of oxide particle size on the VALE Fe3O4 → FeO reaction is shown in Figure 6 for reduction at 1000 °C. The reduction rate is essentially the same for average particles that range from 110 μm to 508 μm in size. This result is a strong indication that gas diffusion through the oxide pore is fast and does not hinder the rate under these conditions. This way, the reducing gas is allowed to diffuse through the pore network before reacting in the pore walls, which is the major reaction resistance which follows the uniform internal reduction model. If gas diffusion through the oxide pores was playing a role either as limited mixed control or product layer diffusion, then the time to achieve a given fractional reduction would be proportional to the initial particle radius (r o ) or particle radius squared (r 2 o ), respectively.

Effect of the particle size on the Fe3O4 → FeO reduction of VALE at 1000 °C
The rate of Fe3O4 → FeO reduction begins to decrease when particles larger than 0.5 mm are employed, which suggests that pore diffusion resistance becomes important as the diffusion distance increases. In addition, it was observed that the effect of particle size on the reduction rate became less pronounced with decreasing reaction temperature, and it was essentially negligible at 700 °C within the VALE size range. Because of the narrow size distribution of PAH and Taconite samples, meaningful evaluation of particle size effect on the reduction of those oxides was not possible.
Apparent Reaction Rate Constants
In theory, the intrinsic rate constant (k) for a specific reduction reaction at a given temperature should be independent of the type and geometry of the iron oxide employed. However, because of the porous nature of the oxide fines used in the present work, determination of the intrinsic rate constants is complex because the actual area participating in the reduction cannot be described precisely. Although the total specific surface area can be measured, it does not necessarily represent the actual reacting area of oxide. As an example, Turkdogan[13] concluded from his work on the reduction of dense and porous wustite to metallic iron that only about 1 to 2 pct of the total pore surface area was used effectively during reduction. Moreover, as the total pore surface area increased, the fraction of pore wall area used decreased.[13] Therefore, instead of k, the product of the reaction rate constant and the pore specific surface area (k·S) is evaluated by applying the appropriate reduction model for each oxide sample studied.
For VALE and Taconite, experimental results as well as the observation of partially reduced samples indicate that the Fe3O4 → FeO reaction follows the uniform internal reduction mechanism. Therefore, Eq. [6] can be applied to the reduction data, and (k·S) is derived from the slope of the curves at each temperature, shown in Figures 7(a) and (b) for VALE and Taconite, respectively.

Uniform internal reaction model applied to Fe3O4 → FeO reduction data at several experimental temperatures, (a) VALE and (b) Taconite
The rate-controlling step for the Fe3O4 → FeO reduction of PAH is more complex. Results indicate that a combination of mechanisms that include gas diffusion and chemical reaction in the oxide pores as well as external gas mass transfer is determining the overall reduction rate in the temperature range studied. Thus, the reduction model developed for spherical particles that include limited mixed control and external gas mass transfer (Eq. [11]) is employed to the PAH reduction data, and results are given in Figure 8. Then, the limited mixed control rate constant (klm) is determined for each temperature. The use of the model was restricted to reduction temperatures below 700 °C. Above 700 °C, external gas mass transfer resistance is the predominant mechanism, and the correction necessary to obtain klm becomes excessively large.

Limited mixed control and external gas mass transfer model applied to PAH from 590 °C to 700 °C
The temperature dependence of the Fe3O4 → FeO reaction rate constants, (k·S) for VALE and Taconite as well as (klm) for PAH, is shown in Figure 9. Considering the temperature range from 700 °C to 1000 °C, the (k·S) varies from approximately 120 to 8500 cm3/mole-O·sec for VALE and from 30 to 2500 cm3/mole-O·sec for Taconite. Despite the higher values of (k·S) for VALE (which is a result of the faster reaction rates of that oxide) the slopes of VALE and Taconite are similar. For PAH, the calculated limited mixed control rate constant (klm) is about 0.7 cm/sec at 700 °C, which is approximately 17 times larger than at 590 °C. The temperature dependence of klm is about the same as (k·S). In theory, the temperature dependence of klm is expected to be about half of the true intrinsic reaction rate constant because it contains the effective diffusivity (D e ), which is not a strong function of temperature. The surprisingly high klm temperature dependence could be the result of the relatively narrow temperature range in which klm was computed. In addition, the pore surface area and, consequently, the effective gas diffusivity may vary with temperature.

Temperature dependency of the reaction rate constant product and specific surface area (k·S) for VALE and Taconite as well as the limited mixed control rate constant (klm) for PAH
It is an interesting exercise to extract (k·S) for PAH from klm. Obtaining the values of (k·S), however, requires knowledge of the oxygen molar density (ρ o ) and the D e . Although the oxygen molar density can be obtained from data in Table I, estimation of the effective diffusivity requires knowledge of the mechanism that dominates the gas diffusion through the pores of PAH during reduction. This determination can be assessed by analysis of the Fe3O4 → FeO reduction by the CO–CO2 gas diluted in Helium or Argon that was previously discussed in Figure 4. Considering limited mixed control (as in Eq. [7]) the reduction rate is expected to be proportional to the square root of the gas diffusivity for specific experimental conditions. The gas diffusivity in the pores of the oxide (D p ) is the result of resistances related to molecular (D m ) and Knudsen (D K ) diffusion.[18–20]
In Figure 4, the Fe3O4→FeO reduction rate of PAH was approximately 1.9 times faster for CO–CO2 diluted in Helium than Argon. Because the average molecular diffusivity of CO–CO2 in Helium (D i-He) is about four times greater than the molecular diffusivity of CO–CO2 in Argon (D i-Ar), it may be concluded from the ratio of the calculated diffusivities in Eq. [14] that molecular diffusion primarily took place through the pores of PAH.
D e can be estimated roughly by the correlation of Weisz and Schwartz[30] in which only molecular diffusion (D m ) is taken into account.
where ε represents the average porosity of Fe3O4 in PAH and is calculated using the particle and true densities (ρ p and ρ t respectively) as reported on Table I, under the assumption that the particle geometry does not change considerably.
With a density (ρ o ) of approximately 0.007 g·atom-O/cm3 and D e of about 0.2 cm2/sec at 700 °C, the PAH (k·S) value was computed to be 300 cm3/mole-O·sec, which is about three and ten times larger than VALE and Taconite, respectively. Considering the reduction rates of the three oxides, the (k·S) of PAH seems somewhat low, which could result from an overestimation of the D e .
Sintering Effect on Reduction
Compared with VALE and Taconite, the reduction rate of PAH was considerably faster, to an extent that the rate limiting step was determined by a mixed regime that comprised external gas mass transfer along with pore diffusion and chemical reaction in the oxide pores. Apart from the impurities content among the oxides, the major variations in reaction rate and limiting step may occur from differences in oxide internal surface area or porosity. Upon heating, the oxide pore surface area may sinter and decrease, which can influence the rate of reduction and possibly the dominating reaction mechanism. More experiments were carried out in which PAH was preheated to temperatures that ranged from 900 °C to 1100 °C before reduction at 620 °C. For comparison, the same procedure also was applied to VALE. The effect of preheating on the Fe3O4 → FeO reduction rate was distinct for PAH and VALE. Although no changes in reduction kinetics were observed for VALE, preheating above 900 °C decreased the PAH reduction rate reacted at 620 °C, as shown in Figure 10.

Effect of preheating temperature on reduction of Fe3O4 → FeO of PAH at 620 °C
This observation could be associated with the greater driving force for the sintering of PAH because of its larger specific surface area.[31] Although the specific surface area was not measured directly after preheating, the change in the reaction rates corresponded to a decrease of available reacting areas as preheating temperature increased above 900 °C. Doherty[22] reported significant changes in the specific surface area of Brazilian hematite fines heated from 300 °C to 800 °C. Compared with the present results, it may be speculated that despite the change in the total specific surface area that may occur, the effective reacting area does not seem to modify extensively until about 900 °C for PAH.
Figure 11 shows the cross section of PAH, preheated to 1000 °C and subsequently partially reduced at 620 °C, when the experiment was interrupted within the Fe3O4 → FeO region. Although a macroscopic receding interface is evident, substantial reaction also occurs ahead of the interface, even at the center of the oxide particle. This observation implies that the reacting gas had more time available to diffuse through the oxide pore structure before reacting in the pore walls, which was a consequence of the PAH reacting area decrease on preheating. As a result, the gas radial concentration profile will differ from the limited mixed control, because equilibrium ahead of the reacting interface is not readily established. This reduction mechanism, therefore, differs from the limited mixed control because the reaction occurs well ahead of the interface. On sintering, PAH becomes denser and comparable with the other commercial oxides (VALE). In addition, the reduction controlling mechanism seems to turn away from limited mixed control to partial internal reduction, which is a transition regime between the former and uniform internal reaction.[16]

Cross-sectional view of partially reduced PAH within the Fe3O4 → FeO region preheated to 1000 °C for 60 minutes before reduction at 620 °C
Summary and Conclusions
The reduction of iron oxide fines to FeO with an equal volume CO–CO2 gas between 590 °C and 1000 °C was investigated. The main findings are summarized as follows:
-
1.
Reduction from Fe2O3 to FeO proceeds in a well defined stepwise mode with Fe3O4 as an intermediate oxide.
-
2.
The first step of reduction (Fe2O3 to Fe3O4) is fast and controlled by external gas mass transfer independent of the temperature and iron oxide type employed in this work.
-
3.
The second reduction step (Fe3O4 to FeO) is slower and is the overall reaction controlling step. The rate-controlling mechanism varies with temperature and iron oxide type. For moderately porous and denser oxide fines, such as VALE and Taconite ores, complete internal reduction takes place across the temperature range and particle sizes that were investigated.
-
4.
For highly porous oxides, such as PAH, the Fe3O4 → FeO reduction is dominated by external gas mass transport through temperatures above about 700 °C for the present experimental conditions. Below 700 °C, a mixed reaction mechanism, which involves external gas mass transfer and limited mixed control, predominates.
-
5.
The product of the intrinsic reaction rate constant and specific surface area (k·S) was evaluated for the Fe3O4 → FeO reduction step for VALE and Taconite with the uniform internal reduction model. (k·S) varied from 120 to 8450 cm3/mole-O·sec and from 30 to 2500 cm3/mole-O·sec for VALE and Taconite, respectively from 700 °C to 1000 °C. At 700 °C, the (k·S) value of PAH was about 350 cm3/mole-O·sec, which is approximately 3 and 12 times larger than the (k·S) obtained for Vale and Taconite, respectively.
-
6.
Sintering of PAH before reduction decreases the overall reaction rates. The Fe3O4 → FeO controlling mechanism seems to shift from limited mixed control toward partial internal reaction.
References
E.T. Turkdogan and J.V. Vinters: Metall. Trans., 1971 vol. 2, pp. 3175-88.
Turkdogan, E.T.: Metall. Trans. B, 1978, vol. 9, pp. 163-79.
R. Corbari and R.J. Fruehan: Carnegie Mellon University, unpublished research, 2004.
R.J. Fruehan, K. Ito, and B. Ozturk: Trans. ISS: 1988, pp. 83–91.
R.J. Fruehan: Trans. ISS, 2003, vol. 30 no. 2, pp. 48-60.
R.S. Sampaio, R.J. Fruehan and B. Ozturk: Trans. ISS, 1993, vol. 14, pp. 59-67.
E.T. Turkdogan: Metall. Mater. Trans. B, 1978, vol. 9, pp. 163-79.
A.A. El-Geassy and M.I. Nasr: Iron Steel Inst. Jpn., 1988, vol. 28, no. 8, pp. 650-8.
H.Y. Sohn and J. Szekely: Chem. Eng. Sci., 1972, vol. 27, no. 4, pp. 763.
S.P. Trushenski, K. Li and W.O. Philbrook: Metall. Trans., 1974, vol. 5, pp. 1149-58.
J. Szekely, J.W. Evans and H.Y. Sohn: Heterogeneous Reaction Kinetics, Academic Press, Washington, DC, 1976.
J. Szekely and J.W. Evans: Metall. Trans., 1971, vol. 2, pp. 1691-711.
E.T. Turkdogan and J.V. Vinters: Metall. Trans., 1972, vol. 3, pp. 1561-74.
R.H. Spitzer, F.S. Manning and W. Philbrook: Trans. Metall. Soc. AIME, 1966, vol. 236, no. 5, pp. 726.
W.M. Mckewan: Trans. Am. Inst. Mining Metall. Eng., 1958, vol. 212, pp. 791–93.
R.H. Tien and E.T. Turkdogan: Metall. Trans., 1972, vol. 3, pp. 2039-48.
S.R. Story and R.J. Fruehan: Metall. Mater. Trans. B, 2000, vol. 31, pp. 43-54.
R.W. Pollard and R.D. Present: Phys. Rev., 1948, vol. 73, pp. 762-74.
G.R. Youngquist: Flow Through Porous Media, American Chemical Society, Washington, DC, 1970
A. Wheeler: Reaction Rates and Selectivity in Catalyst Pores, in Advances in Catalysis and Related Subjects, Academic Press, Washington, DC, 1951.
T. Murayama, Y. Ono and Y. Kawai: Iron Steel Inst. Jpn., 1977, vol. 63, no. 7, pp. 1099-107.
R.D. Doherty, K.M. Hutchings, J.D. Smith and S. Yörük: Metall. Mater. Trans. B, 1985, vol. 16, pp. 425-32.
H.Y. Sohn: Chem. Eng. Sci., 2004, vol. 59, pp. 4361-68.
K. Piotrowski, K. Mondal, T. Wiltowski, P. Dydo and G. Rizeg: Chem. Eng. J., 2007, vol. 131, no. 1-3, pp. 73-82.
T. Ariyama, S. Isozaki, S Matsubara, H. Kawata, K. Kondo and I. Kobayashi: J. Iron Steel Inst. Jpn., 1993, vol. 79, no. 12, pp. 1323-28.
N. Towhidi and J. Szekely: Metall. Trans. B, 1983, vol. 14, pp. 359-67.
D. Wagner, O. Devisme, F. Patisson, and D. Ablitzer: Sohn International Symp., San Diego, CA, 2006.
N.A. Warner: Trans. Metall. Soc. AIME, 1964, vol. 230, pp. 163-76.
R. Corbari: Ph.D. Thesis, Carnegie Mellon, Pittsburgh, PA, 2008.
P.B. Weisz and A.B. Schwartz: J. Catal., 1962, vol. 1, pp. 399-406.
R.M. German: Sintering Theory and Practice, Wiley, New York, NY, 1996.
Acknowledgments
The authors wish to thank the member companies of the Center for Iron and Steelmaking Research for the funding of this research. Iron oxide samples provided by Vale and the U.S. Steel Technology Center as well as laboratory analysis by Vesuvius Research also are appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted March 8, 2009.
APPENDIX: External Gas Mass Transfer Determination
APPENDIX: External Gas Mass Transfer Determination
The mass transfer coefficient of CO–CO2 (mCO-CO2) that was used to compute the reduction rate associated with external gas transfer was determined indirectly by measuring the evaporation rate of Mg under Argon-5 pct H2 and H2 at 625 °C. The equipment, flow and geometric conditions of the evaporation experiments were identical to those conditions of the iron oxide reduction. The results are shown in Figure A1.

Weight loss of Mg powder as a function of time at 620 °C under a flow of 5 lpm of Ar-5 pct H2 and H2
The evaporation rate of magnesium in Argon (RMg-Ar), which is controlled by external gas phase mass transfer, is expressed in Eq. [A1], where mMg-Ar is the mass transfer coefficient of Mg in Argon, and p SMg and p BMg represent the partial pressure of Mg in the surface and bulk stream, respectively.
Using Eq. A1, the mass transfer coefficient (m Mg-Ar) is calculated with the experimental evaporation rate and by taking Aδ as the crucible area; \( p_{\rm Mg}^{S} \) is calculated as the equilibrium vapor pressure of Mg, whereas \( p_{\rm Mg}^{B} \) is assumed to be negligibly small. The mass transfer coefficient of Mg–Ar at 625 °C then is extrapolated to the temperature range of interest (600 °C–1000 °C) and converted to mass transfer of CO–CO2 which accounts for the differences in gas diffusivities. This methodology was validated using the Mg–H2 evaporation results also shown in Figure A1.
Rights and permissions
About this article
Cite this article
Corbari, R., Fruehan, R. Reduction of Iron Oxide Fines to Wustite with CO/CO2 Gas of Low Reducing Potential. Metall Mater Trans B 41, 318–329 (2010). https://doi.org/10.1007/s11663-009-9315-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-009-9315-2