
Abstract
The formation and development of the mineral layer that forms between coke and liquid iron during carbon dissolution has been characterized. Coke particles (−2 mm, +0.5 mm) were added to the top surface of an iron 2 mass pct C melt at representative iron-making temperatures, for periods of time between 2 and 120 minutes, before being quenched. The quenched samples were then sectioned, and the solidified coke-melt interfacial region analyzed in the scanning electron microscope (SEM). Analysis showed that a mineral layer was present at the interface at all experimental temperatures (1450 °C to 1550 °C) from 2 minutes and persisted beyond 120 minutes. The mineral layer was found to be composed of calcium aluminate phases, with the proportions of these phases dictating its morphology. Further, changes observed in the rate of carbon dissolution from the coke were related to the composition and morphology of the mineral layer. The effect of this mineral layer on the rate of carbon dissolution has been interpreted as a change in the reaction control mechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coke is the primary solid material remaining at the level of the hearth and throughout the lower zones of the blast furnace. It is the principal source of fuel for the furnace and provides mechanical support for the burden above it. Additionally, coke supplies the carbon required for the carbonization of the liquid hot metal in the hearth.[1] This investigation is focused on the coke-metal reactions occurring in the hearth below the slag layer. Liquid iron entering this region contains more than 2 mass pct C and picks up its final carbon while percolating through the packed-coke bed in the deadman and hearth of the blast furnace.[1]
Metallurgical coke typically contains 8 to 12 pct by mass inorganic mineral matter,[2] derived from the mineral matter of the parent coals.[3,4] As coke is dissolved in the liquid iron, there is potential for the insoluble components of this inorganic mineral matter to form a layer at the surface of the coke, inhibiting carbon dissolution.[5–12]
Previous Work
There is a significant body of research that has focused on the kinetics of coke dissolution into iron.[6,10–11,13–18] Coke dissolution into iron is generally considered to be controlled by first-order kinetics and is frequently described by the rate of carbon dissolution if liquid side mass-transfer limits the dissolution reaction, as presented in the following equations:
Under the assumption of ideal mixing within the melt, the mass balance for carbon can be integrated (assuming A, V, and k m are all time independent) as follows:
where J is the flux (composition·m·s−1), D is the diffusion coefficient of carbon in liquid iron (m2·s−1), δ is the effective boundary-layer thickness (m), [C]sat is the carbon concentration at carbon saturation (mass pct), [C]bulk is the bulk-carbon concentration in melt (mass pct), [C]0 is the initial carbon concentration of bulk at t = 0 (mass pct), V is the volume of melt (m3), A is the area of reaction interface (m2), t is the time (s), and k m is the mass-transfer coefficient (m·s−1).
The generic term, “ash,” as used in the iron-making literature, can sometimes be misconstrued as the mineral-matter residue from coke combustion (reaction with a gas phase), as opposed to a residual product of coke-liquid reactions, as in the case for coke dissolution in liquid iron. In this article when discussing our results, the inorganic and mineral components of coke will be referred to as mineral matter. To ensure there is no misrepresentation of other researchers’ work, when referring to their work, the term, ash, will continue to be used.
Several researchers have reported on the role ash plays in reducing the rate of carbon dissolution into iron from coke.[6,10,17] Orsten and Oeters[10] reasoned that a solid-ash product would be distributed across the reaction surface, reducing the available contact area between the carbonaceous material and the melt; however, if the ash product was liquid, it could be flushed away from the interface, reducing this effect.
Gudenau et al.[6] indicated that an ash film formed on the surface of the coke and that the presence of this film was significant in reducing the rate of carbon dissolution from the coke. This study also found that the addition of refractory oxides, CaO, MgO, and Al2O3, to the coke further decreased the dissolution rate, while additions of iron oxide enhanced the dissolution rate.
Following experiments that used a sessile-drop apparatus to react a drop of iron with a carbonaceous substrate, Wu et al.,[19] McCarthy et al.,[8–9] and Khanna et al.[7] reported on the presence and composition of the ash product at the droplet/carbonaceous-material interface. General observations of the droplet surface in these studies indicate that although silica was contained in the carbonaceous material, there was none present in the ash at the interface. The ash at the interface was initially Al2O3; however, as the reaction time increased, the proportion of CaO increased, after which the Al2O3 was observed to disappear from the droplet-coke interface and was replaced by an iron-calcium sulphide. Although these studies are instructive in identifying potential features of a mineral-matter layer at the metal-coke interface, the coke-metal mass ratios used are not typical of those found in the blast furnace. In the hearth of the blast furnace, the coke is immersed in the iron below the slag line. In a sessile-drop experiment, a small finite amount of iron sits on a substrate of coke or carbon. Under such conditions, it would be expected that the coke-metal ratio of the blast furnace would be much lower, with the potential for silicon and sulfur levels of the metal, as a result of coke dissolution, to also be much lower. This would have a significant impact on any mineral layer formed at the interface.[5]
Khanna et al.[7] also observed a significant change in the rate of carbon dissolution over time in some chars. Chars with high CaO levels displayed a two-stage behavior, whereby after a period of time, the rate of dissolution would decrease significantly. This two- stage behavior was attributed to the increased deposition of calcium-based reaction products blocking the reaction surface.
In a recent study by the current authors,[5,12] examination of the coke-iron interface of coke samples immersed in liquid iron showed that the type of mineral layer formed was temperature dependent. No mineral layer was observed at 1550 °C, while at lower temperatures (1500 °C to 1400 °C), a mineral layer was observed. The formation of the layer was described in terms of a temperature-activated time-dependent sintering/fusion mechanism. In this study, it was also found, consistent with the work of Wu et al.,[19] McCarthy et al.,[8,9] and Khanna et al.,[7] that SiO2, although the largest single component of the mineral matter in the unreacted coke, was essentially absent from the coke-iron mineral layer.
Husslage,[20] in an excellent study of liquid (iron and slag) flowing through a packed- coke bed, found that iron and slag flow concurrently, generally in rivulets that are not evenly distributed through the bed. Husslage[20] characterized a range of (macro-scale) interactions observed to have occurred between the coke, slag, and iron draining through a coke bed. General findings of this work include that slags were observed to have wetted the coke better than the iron (iron–5 mass pct C close to carbon saturation). Slags that wetted the coke were able to flow into the cokes pores and that metal with a lower carbon level (iron – C 3.8 mass pct) was observed to have wetted the coke surface and was able to penetrate the pore structure. This study also indicated that where slag had entered the coke porosity, there was evidence of silica reduction and transfer of silicon to the iron.
There has been no definitive study published in the literature that has characterized the development of the mineral-matter layer with time and temperature and its affect on the coke (carbon) dissolution. Characterizing the development of the mineral layer is the focus of this study and the experimental results presented in this article.
Experimental
A series of coke (carbon)-dissolution experiments were conducted, where 35 g of crushed coke (−2 mm, +0.5 mm) were added directly to the top surface of 572 g of liquid-iron 2 mass pct carbon alloy. The melt was frequently sampled with a 1-mm ID quartz tube over a period of 2 hours. The experiment was carried out in a dry argon atmosphere (>99.997 pct purity). The argon-gas flow rate during the experiment was 0.72l/min. These experiments were conducted over the temperature range of 1450 °C to 1550 °C. The temperatures were chosen to replicate what might be expected in the lower-zone deadman area of a blast furnace.
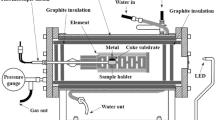
A second series of quenched coke (carbon)-dissolution experiments were performed, where 10 g of crushed coke (−2 mm, +0.5 mm) were added directly to the top surface of 164 g of liquid-iron 2 mass pct carbon alloy. The melt, plus coke, were held at temperature for a period of time ranging from 2 to 120 minutes. At the end of this time, the crucible containing the melt and coke was quenched by lowering it into a water-cooled stainless-steel quenching chamber fitted to the bottom of the furnace. The experiment, including the quenching, was carried out in a dry argon atmosphere (>99.997 pct purity). The argon-gas flow rate during the experiment was 0.94l/min and was increased to 9.4l/min during quenching. After quenching, the samples were sectioned and prepared for electro-optical examination. Experiments were conducted over the same temperature range of 1450 °C to 1550 °C as in the coke-dissolution series. A schematic of the furnace and sample configuration is given in Figure 1. A schematic comparison of the samples used in both series of experiments is given in Figure 2.

Schematic of the furnace configuration used in the quenched-coke dissolution experiments

Schematic showing experimental samples used for (a) quenched coke dissolution series and (b) coke dissolution series
Carbon and sulfur analysis was performed using an LECOFootnote 1 CS-444 analyzer. Analysis of other elements in the iron was performed by atomic emission spectroscopy, at the metallurgical laboratories of BlueScope Steel, on disks of iron machined from the middle of the iron samples.
The iron mass used in the dissolution series was limited by crucible volume. The coke mass was determined such that if sufficient carbon was transferred to the iron melt for it to become carbon saturated, there would be a 10-mm layer of coke on the iron surface to maintain an excess of carbon. The crucible, iron mass, and coke masses used in the quenched series of experiments were determined to maintain the key geometric factors of coke-metal mass ratio and melt mass-surface area ratio. These values are given in Table I.
Raw Materials
The iron-carbon alloy was prepared in-situ before the carburizer was added by melting appropriate amounts of electrolytic iron and coarsely crushed spectrographic-grade graphite rod to achieve a 2 pct carbon-iron alloy prior to the addition of the coke. Melting was achieved by heating the iron and graphite to 1540 °C for 10 minutes before heating/cooling to the experimental temperature. The consistency of the initial alloy composition was monitored and found to be 2.03 ± 0.05 mass pct C and independent of the experimental temperature. The initial melt composition is presented in Table II. Carbon loss/pickup from the melt to the furnace atmosphere was monitored by a series of runs with no carburizer added. In these tests, the variation in carbon level was found to be +/− 0.025 mass pct C at any point over a 2-hour period.
The coke samples used were provided by BlueScope Steel and contained approximately 0.4 to 0.45 mass pct sulfur and 11.6 mass pct inorganic material. The inorganic material can be considered to be refractory in nature. A detailed oxide composition measured by X-ray fluorescence (XRF) after ashing at 815 °C is given in Table III.
The lump coke was coarsely crushed to −20 mm in a jaw crusher, then further crushed in a roll crusher to obtain the desired −2 mm, +0.5 mm size fraction used. The −2 mm maximum size was selected to minimize any effects on coke dissolution caused by the presence of large cracks and fissures present in coke, while the −0.5 mm minimum size prevents the coke from being entrained in the furnace-exhaust gas stream when added.
Quenched Sample Preparation
The crucibles containing the iron alloy and coke from the quenched experiments were impregnated with liquid resin under vacuum to preserve the coke-metal interface during sectioning. Lead shot was added to the top of the coke inside the crucible, before the resin impregnation, to minimize coke movement during the vacuum-impregnation procedure. The bottom section of the crucible was removed, and iron samples for analysis were machined from the iron block. A vertical cut was made across the center of the sample, exposing the coke-metal interface. The sample was mounted in epoxy resin and prepared for SEM analysis by grinding with P500 SiC paper, followed by three polishing steps, using 9-, 3-, and 1-μm diamond.
Assessment of Interfacial Mineral Layer
Scanning electron microscope analysis, involving electro-optical analysis, X-ray mapping, and energy dispersive spectroscopy (EDS) analysis, was performed over large areas of the coke-iron interface from the quenched series of experiments. The results reported from this analysis are observations and measurements obtained from the quenched series of experiments and of the coke-iron interface after quenching. Unless otherwise stated, it is assumed that the results reported and discussed are representative of the high-temperature phenomena. Where mineral-matter compositions are reported, they are derived from EDS analysis of the quenched samples.
Results
Carbon Transfer
Carbon pickup for the quenched and nonquenched dissolution experiments is plotted for 1450 °C, 1500 °C, and 1550 °C as a function of time in Figure 3. The dashed line presented in Figure 3 represents both the quenched and nonquenched data sets. There is good agreement in the level of carbon pickup with time between the two experimental methods employed in this study. This agreement indicates that the same factors that affect the kinetics of carbon transfer from coke to the iron are active in both sets of experiments.

[C] vs time: (a) 1450 °C, (b) 1500 °C, and (c) 1550 °C
At all temperatures, there is an initial period of unstable carbon pickup occurring in the first 6 to 8 minutes of the dissolution experiments. This initial instability is likely to be contributed to by many factors, such as cooling of the melt when the cold coke is added, air/oxygen entrained with or present in the coke porosity, and disturbance of the melt surface during the frequent initial sampling.
In the case of the 1450 °C and 1500 °C quenched carburizer cover results, the carbon level is seen to increase steadily before reaching a maximum of approximately 3.0 and 3.6 mass pct at 60 and 40 minutes, respectively. Beyond this, there was little subsequent carbon pickup in the melt. In the case of the 1550 °C quenched carburizer cover results, the carbon pickup was initially rapid, and although no clear plateau was observed, the rate of carbon transfer to the melt slowed. This rate reduced over the experimental time with carbon reaching a maximum of approximately 4.67 mass pct after 120 minutes of contact. The maximum carbon levels reached in these experiments are significantly below the carbon- saturation levels calculated using the thermodynamic software package MTDATA.[21] Melt compositions used to calculate the carbon-saturation values are based on the average values beyond 60 minutes for 1450 °C and 1500 °C, and beyond 90 minutes for 1550 °C. These average melt compositions and the calculated carbon-saturation values are given in Table IV.
Layer Formation
Typically, evidence of a mineral layer was observed in all samples at all experimental times from 2 to 120 minutes over the three experimental temperatures of 1450 °C, 1500 °C, and 1550 °C. Due to the large numbers of samples and the large areas examined, only selected representative images are reported. Upon cooling, the iron-carbon alloys experience a significant increase in density, resulting in contraction of the melt surface away from the original iron-coke interface. This contraction of the melt results in either a gap between the melt and the original mineral layer or in the distortion of the original mineral layer as it adheres to the contracting iron surface. A typical section of quenched interface after five minutes at 1500 °C is presented in Figure 4. The mineral layer is observed following the profile of the iron surface, although the original liquid-iron surface has contracted away from the mineral layer. The resulting voids are filled with resin during the metallographic preparation of the sample.

QBSD SEM images of the quenched coke-mineral layer-iron interface, quenched after 5 min at 1500 °C
The composition of the mineral layer was found to be principally composed of calcium aluminates. The ratios of calcium and aluminum found in the mineral layer indicate that the mineral layer is composed of alumina and the calcium aluminates, CaO.6Al2O3, CaO.2Al2O3, and CaO.Al2O3. The melting points of these phases are 2053 °C, 1830 °C, 1762 °C, and 1602 °C,[22] respectively. The melting points of these phases are well above the experimental temperatures. Microscopic investigation revealed that no rounding of the sharp edges and points of the mineral matter at the interface, as may be expected if a liquid phase had been present, was observed. Additionally, a liquid phase could be expected to collect or pool in the center or at the edge of the iron-coke interface on cooling. Such a pool of material was not found in any of the samples sectioned and examined. Therefore, it is assumed that the original mineral layer was solid before quenching and that the original solid structure and composition of the mineral layer is retained in the quenched sample.
The amount of material present in the mineral layer between the coke and liquid iron was observed to increase with reaction time. The morphology of this layer was also observed to change with time, transforming from an initial loose agglomeration of particles, through an acicular open structure, to a dense layer. The level of calcium in the calcium aluminate was found to increase with both reaction time and temperature.
The initial stages of the formation of the mineral layer can be observed in Figure 5 and Figure 6. In Figure 5, a partially dissolved coke particle is present near the melt interface. Although the iron has contracted away from the surface of the coke particle on cooling, the coke-iron interface is still evident. On the coke side of the interface, the original mineral matter (primarily made up of aluminosilicates) is contained in a carbon matrix. On the melt side of the interface, mineral matter that was exposed as the carbon matrix dissolved and accumulated at the melt surface. The accumulated material forms an agglomeration of loosely packed particles, primarily composed of Al2O3. Figure 6, a higher resolution image of the Al2O3 layer, shows the layer has an open-pore structure.

QBSD SEM image and elemental X-ray maps of coke particle on the interface quenched after 2 min at 1500 °C. Numbered regions have been analysed using EDS, with the compositions given in Table V

QBSD SEM image and elemental X-ray maps of mineral layer quenched after 2 min at 1500 °C
Figure 5 also illustrates the behavior of silica at the coke-iron interface. Silica is regularly observed in undissolved coke particles present near the coke-iron interface; however, it is generally absent from the mineral layers that are formed at the coke-iron interface. This absence of SiO2 from the mineral layer at the coke-iron interface is attributed to reduction of the SiO2 by solute carbon in the melt. The reduction of SiO2 to [Si] is supported by the increase in the silicon levels in the melt from the initial melt composition as presented in Table IV. Silica reduction from coke and the mineral layer has been discussed in detail previously.[5]
Development of the Mineral Layer
As the coke dissolution reaction continues, the predominant structure changes from a loose agglomeration of primarily alumina particles to an open porous network of acicular needles (Figures 7 and 8). These needles are still predominately Al2O3 but also contain an increased proportion of calcium, up to approximately 9 mass pct. Further dissolution increases the calcium enrichment of the mineral layer. This layer retains a relatively open structure; however, the fine needles evident in Figures 7 and 8 are replaced by a coarser structure (Figure 9). The composition of the mineral layer has also continued to change, with CaO comprising approximately 20 mass pct of the layer. As the dissolution-reaction time continues, further calcium enrichment of the mineral layer is observed, with CaO levels reaching in excess of 26 mass pct. The structure of the predominant mineral layer changes from being an open structure (Figures 6 through 9) to a dense layer that is well bonded to the iron surface (Figure 10).

QBSD SEM image and elemental X-ray maps of mineral layer quenched after 5 min at 1450 °C. Numbered regions have been analyzed using EDS, with the compositions given in Table V

QBSD SEM image and elemental X-ray maps of mineral layer quenched after 2 min at 1500 °C

QBSD SEM image and elemental X-ray maps of mineral layer quenched after 60 min at 1450 °C. Numbered regions have been analysed using EDS, with the compositions given in Table V

QBSD SEM image and elemental X-ray maps of mineral layer quenched after 60 minutes at 1500 °C. Numbered regions have been analysed using EDS, with the compositions given in Table V
The EDS analysis for regions indicated by numbered rectangles in Figures 5, 7, 9, and 10 are given in Table V. Molar ratios of CaO to Al2O3 are given for analysis regions of the coke-iron interfacial mineral layer. Where sulfur was present, it has been attributed to the CaS phase and the amount of CaO reduced accordingly.
The role of calcium enrichment in the mineral layer at the melt interface can be understood by considering the CaO-Al2O3 binary phase-equilibrium diagram (Figure 11).[22] There are three distinct calcium aluminates at the alumina-rich end of the phase diagram. The structures of these calcium aluminates have been well characterized as they are important with respect to cleanness in the steelmaking process.[23] Key details of these calcium aluminates are given in Table VI.

CaO-Al2O3 phase diagram reproduced from Slag Atlas[22]
It should be noted that the authors are not suggesting that the calcium aluminates formed during coke dissolution have any role to play in cleanness issues later in the steelmaking process.
The calcium-aluminate structures described in Table VI are evident in the micrographs of the mineral layer presented in Figure 6 to Figure 10. The EDS analysis of the needlelike structures in Figure 7 and Figure 8 show that the molar CaO/Al2O3 ratio is 0.19, indicating that the mineral layer is predominantly the calcium aluminate, CA6. The needlelike acicular structure shown in Figures 7 and 8 is typical of CA6.
The EDS analysis in the regions indicated in Figure 9 shows a molar CaO/Al2O3 ratio of 0.44 to 0.46. Such a ratio indicates a mixture of the calcium aluminates, CA2 and CA6, with the mixture consisting of approximately 85 pct of the CA2 phase (as determined from Figure 11). Such a structure could be expected to retain some of the fine acicular CA6 needles but would also contain significant amounts of the larger, 2-D plates of CA2 crystals. This is consistent with what is observed in Figure 9.
The dense calcium-aluminate structure shown in Figure 10 has a molar CaO/Al2O3 ratio of 0.67. This ratio indicates that the mineral-matter layer contains approximately 66 pct of the CA2 phase with the remaining 34 pct being the CA phase (Figure 11). The CA phase is a dense fine-grained phase and has developed around the CA2 phase, resulting in densification of the mineral-matter layer.
Figure 12(a) details the development of the coke-iron mineral layer in terms of its molar CaO- Al2O3 ratio. From Figure 12(a), it can be seen as the calcium level in the mineral layer approaches a plateau. The time to reach this plateau decreases with increasing temperature. Also, the level of calcium present in the mineral layer at the plateau increases with increasing temperature.

(a) Molar CaO/Al2O3 ratio vs experimental time and (b) predominant phase at the melt interface vs experimental time
Combining the information presented in Figure 12(a) with the CaO-Al2O3 phase diagram (Figure 11), the predominant calcium-aluminate phases present in the coke-iron interfacial mineral layer can be determined (Figure 12(b)).
Coke Dissolution Kinetics
Coke dissolution into iron is considered to be a first-order kinetic process and is generally described as being limited by liquid-phase mass transfer of carbon in iron as given in Eq. [2]. The mass-transfer coefficient, k m , for such a process can be evaluated from the slope of a plot of \( \frac{V} {A}\ln {\left( {\frac{{[C]_{{{\text{sat}}}} - [C]_{0} }} {{[C]_{{{\text{sat}}}} - [C]_{{{\text{bulk}}}} }}} \right)} \) vs time (Figure 13). The dotted lines in Figure 13 represent linear regions of the plot, the slopes of which are graphical representations of the rate constant, k m . The volume of the melt was taken to be 2.38 × 10−5 cubic meters (based on an assumed density of 6885 kg/cubic meters, corresponding to a Fe-C 2 mass pct alloy at 1500 °C[24]), and the area term was assumed to be the cross-sectional area of the crucible.

First-order mass-transfer control plots for (a) 1450 °C, (b) 1500 °C, and (c) 1550 °C
In Figure 13, it can be seen that there is a significant change in k m , the rate constant, after a period of time for all three temperatures. The change in k m , a decrease in the slope, represents a slowing of the rate of coke (carbon) dissolution into the iron. At the lower temperatures of 1450 °C and 1500 °C after approximately 60 and 50 minutes, respectively, the dissolution reaction effectively stops. At 1550 °C, although the change in the rate constant is not as pronounced, it still represents a significant slowing of the carbon dissolution reaction. The change in k m has been interpreted as a change in the kinetic regime controlling the dissolution of carbon from the coke. It is likely that such a change in the kinetic regime is a continuous change from one regime to another over time, rather than abrupt change.
Although presented here as first-order mass-transfer plots, the data have also been analyzed as first-order chemical control and second-order kinetics. The change in slope with respect to the rate constants was also evident in these approaches.
Effects of Structure on Kinetics
The change in the rate constant is coincident with the change in the predominant phases present in the mineral layer at the coke-iron interface from CA2/CA6 to CA/CA2. Accompanying this change in phase, there is a change in the morphology of the mineral layer from an open acicular structure to a more densely packed structure. This change in the morphology reduces the contact between the carbon in the coke and the liquid iron, and therefore slows the rate of coke (carbon) dissolution.
Consistent with other reported studies in this area,[7–10,13–18] a constant-area term has been used for the contact between the coke and metal. In reality, in the experiments there are two distinct coke-iron contact regimes that may be characterized by two different area terms (or a continuous change in area from a relatively high value to a relatively low value relating to the open and dense structures of the mineral layer, respectively). In constraining the area term to one value, two mass-transfer coefficients (k m ) have been obtained. It is likely that if the contact areas could be determined, the two k m terms would reduce to a single value representing the overall rate of reaction. However, it is not possible to evaluate the contact area accurately in these types of experiments.
Other researchers have inferred that the nature of the mineral layer that could form at the coke-iron interface will affect the kinetics of the carbon-dissolution reaction.[6–9,17,19] The results of this investigation demonstrate for the first time that the composition and subsequent morphology of the mineral layer formed affects the carbon-dissolution reaction.
In the absence of a mineral (product) layer, carbon dissolution into iron is considered to be a first-order liquid-phase mass-transfer process.[6,10–11,13–18] In stating that the nature of the mineral layer that is formed during the coke dissolution affects the kinetics of the carbon dissolution reaction, a change in the reaction control mechanism is implied. On the assumption that mass transfer of carbon in the liquid iron is still in part rate controlling, the formation of the mineral (product) layer provides an additional controlling mechanism. This represents a change from simple mass-transfer control to a mixed-control regime, where both mass transfer and the mineral (product) layer are active. Further, the changing nature of the mineral layer from an open (CA6/CA2) to dense (CA2/CA) structure could also be interpreted as a change in the reaction-control mechanism.
Calcium Enrichment of the Mineral-Matter Layer
Calcium enrichment of the mineral matter at the melt interface is a key component of the formation of the mineral layer and its subsequent densification. As calcium oxides are not readily reduced in liquid iron and steel, and calcium has very little solubility in liquid iron,[25] it can be expected that calcia products will build up at the coke-iron interface as coke dissolves. However, the calcium-enrichment levels are well above what should be expected solely from coke dissolution. Calcium levels in the iron melt have been measured throughout the experiment and were found to have changed from 24 ± 1 ppm calcium initially to 22 ± 1 ppm calcium after 120 minutes. A change in 2.6 ppm calcium of the iron melt would be significant with respect to calcium enrichment of the mineral layer and represent a 10 pct increase in the molar CaO/Al2O3 ratio of the mineral layer. Other workers have reported calcium mobility within coke,[4,26,27] while calcium enrichment at the melt interface has been reported by other researchers.[5,7–8,12,19] This offers the possibility that enrichment of the mineral layer is also a result of calcium mobility within the coke. From the results reported, it is not possible to distinguish between calcium in the mineral layer originating from the coke and the calcium originating from the iron melt. This will form the basis of follow up investigations in the coke-dissolution area.
Wettability
The wettability and interfacial tension of the liquid iron at the mineral-layer interface may also influence the development and growth of mineral layer and its effect on coke dissolution. This influence can be expected both in terms of the structure the mineral-layer forms and penetration of the liquid iron through the mineral layer. These issues relating to wettability and interfacial tension of liquid iron on calcium aluminates formed at the coke-iron interface form the basis of further study in this area.
Application to the Blast Furnace
A mineral layer at the coke-iron interface could be expected to form as coke dissolves into liquid iron.[5–14,17] The question of whether the layer persists and develops at the interface is an important one. If the mineral layer comes into contact with blast-furnace slag, it is likely the layer will be dissolved or modified by the slag. If the mineral layer is formed in the absence of a slag phase, as would be the case in the blast-furnace hearth, beneath the slag level, the mineral layer could persist and develop. Should the coke present in the hearth have been impregnated or coated with slag, as the work of Husslage[20] indicates, the composition and morphology of the mineral formed on coke dissolution would in part be based on the slag composition and the coke mineralogy.
While percolating through the packed-coke bed in the deadman and hearth of the blast furnace, liquid iron picks up its final carbon.[1] Given the slowing of the coke-dissolution rate as a result of the formation of and changes in the mineral layer, it can be expected that the formation of this layer will affect carbon pickup in the blast furnace.
Conclusions
A series of experiments has been carried out in an attempt to describe and characterize the mineral layer formed at the coke-metal interface as the coke dissolved into the liquid iron. The following results were found.
-
1.
The kinetics of carbon dissolution from the coke to the liquid iron were dependent on the structure of the interfacial mineral layer.
-
2.
The changes in the morphology of the mineral layer were directly related to compositional changes, where calcium enrichment of the mineral layer dictated the predominant phase and thus morphology of the mineral layer.
-
3.
The mineral (product) layer formed at the coke-iron interface during coke dissolution is at least in part rate controlling the coke (carbon)-dissolution reaction for the coke-iron system studied.
-
4.
The amount of mineral matter present in the mineral layer was observed to increase with increased reaction time.
-
5.
The composition of the mineral layer was principally composed of oxides of aluminum and calcium, present as various calcium aluminates.
-
6.
The morphology of the mineral layer was observed to change with time, transforming from an initially loose agglomerate of insoluble particles to acicular, before developing into a dense layer.
-
7.
Open structures associated with the alumina-rich calcium aluminates did not appear to significantly inhibit the dissolution of carbon from the coke, while the dense CA phase inhibited dissolution, effectively blocking the dissolution reaction at 1450 °C and 1500 °C.
Notes
LECO is a trademark of LECO Corporation, St. Joseph, MI.
References
Blast Furnace Phenomena and Modelling, Y.E. Omori, ed., Elsevier Applied Science, London, 1987, pp. 57–58
P. Loison, P. Foch, A. Boyer: Coke Quality and Production, Butterworth & Co. Press, London, 1989, p. 159
V. Croft: Ironmaking and Steelmaking, 1980, pp. 116–22
S. Gornostayev, O. Kerkkonen, J. Harkki: Iron Steel Inst. Jpn. Int., 2005, vol. 45, pp. 1–7
M.W. Chapman, B.J. Monaghan, S.A. Nightingale, R.J. Nightingale, and J.G. Mathieson: Iron Steel Inst. Jpn. Int., 2007, pp. 973–81
H.W. Gudenau, J.P. Mulanza, D.G.R. Sharma: Steel Res., 1990, vol. 61, pp. 97–104
R. Khanna, F. McCarthy, H. Sun, N. Simento, V. Sahajwalla: Metall. Mater. Trans. B, 2005, vol. 36B, pp. 719–29
F. McCarthy, R. Khanna, V. Sahajwalla, N. Simento: Iron Steel Inst. Jpn. Int., 2005, vol. 45, pp. 1261–68
F. McCarthy, V. Sahajwalla, J. Hart, N. Saha-Chaudhury: Metall. Mater. Trans. B, 2003, vol. 34B, pp. 573–80
S. Orsten, F. Oeters: Process Technology Proc., Iron and Steel Society, Washington DC, 1986, pp. 143–55
C. Wu, R. Wiblen, and V. Sahajwalla: Belton Symp. 2000, Sydney, Australia, 2000, pp. 409–17
M.W. Chapman, B.J. Monaghan, S.A. Nightingale, R.J. Nightingale, J.G. Mathieson: Australia-China-Japan Symp. Iron and Steelmaking, Liaoning Science and Technology Publishing House, Shenyang, China, 2006, pp. 90–96
S.T. Cham, V. Sahajwalla, R. Sakurovs, H. Sun, M. Dubikova: Iron Steel Inst. Jpn. Int., 2004, vol. 44, pp. 1835–41
S.T. Cham, R. Sakurovs, H. Sun, V. Sahajwalla: Iron Steel Inst. Jpn. Int., 2006, vol. 46, pp. 652–59
S.O. Ericsson, P.O. Mellberg: Scand. J. Metall., 1981, vol. 10, pp. 15–18
N.W. Jones: Ironmaking Steelmaking, 1998, vol. 25, pp. 460–65
M.B. Mourao, G.G.K. Murthy, J.F. Elliott: Metall. Mater. Trans. B, 1993, vol. 24B, pp. 629–38
S. Orsten, F. Oeters: W.O. Philbrook Memorial Symp. Proc., Iron and Steel Society, Toronto, 1988, pp. 27–38
C. Wu, R. Wiblen, V. Sahajwalla: Metall. Mater. Trans. B, 2000, vol. 31B, pp. 1099–104
W.M. Husslage: Ph.D. Dissertation, Delft University of Technology, Delft, 2004, pp. 128–44
R.H. Davies, A.T. Dinsdale, J.A. Gisby, S.M. Hodson, and R.G.J. Ball: Conf. Applications Thermodynamics on the Synthesis and Processing of Materials, Rosemont, IL, 1994, ASM/TMS, Warrendale, PA, 1994, pp. 371–84
M. Kowalski, P.J. Spencer, and D. Neuschutz: in Slag Atlas, VDE (VDEh), ed., Verlag Stahleisen GmbH, Dusseldorf, Germany, 1995, p. 39
R. Kiessling and N. Lange: Nonmetallic Inclusions in Steel, The Metals Society, London, 1978, vol. 2, pp. 36–48
J. Markert and Potschke: Arch Eisenhuttenwes., 1979, vol. 50, pp. 53–56
F. Oeters: Metallurgy of Steelmaking, Verlag Stahleisen GmbH, Dusseldorf, Germany, 1994, pp. 98–101
O. Kerkkonen: Coke Mak. Int., 1997, vol. 9, pp. 34–41
O. Kerkkonen, E. Mattila, R. Heiniemi: 55th Ironmaking Conf. Proc., Iron & Steel Society Ironmaking Division, Pittsburgh, PA, 1996, pp. 275–81
Acknowledgments
The authors thank BlueScope Steel Ltd. and the Australian Research Council for supporting this research. They also thank Dr Huijin Li, Senior Research Scientist, Australian Nuclear Science and Technology Organisation (ANSTO), for access to and assistance using ANSTO SEM facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted on September 18, 2007.
Rights and permissions
About this article
Cite this article
Chapman, M.W., Monaghan, B.J., Nightingale, S.A. et al. Formation of a Mineral Layer during Coke Dissolution into Liquid Iron and Its Influence on the Kinetics of Coke Dissolution Rate. Metall Mater Trans B 39, 418–430 (2008). https://doi.org/10.1007/s11663-008-9145-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-008-9145-7