
Abstract
Hot corrosion behavior of two Ni-based single crystal superalloys containing 0 Re and 2 wt pct Re in molten sodium sulfate (Na2SO4) at 900 °C in static air has been investigated using the “deposit recoat” method. It was shown that the incubation period of the Re-containing alloy was much longer than that of the Re-free alloy. It was because Re increased the stability of the chromium oxide (Cr2O3) scale, promoted the formation of nickel titanate (NiTiO3), and suppressed the diffusion of Ni. In addition, the overall corrosion kinetics of the Re-containing alloy followed a multistage parabolic law, compared with the Re-free alloy. This could be because Re increased the activity of chromium (Cr) and titanium (Ti), which resulted in the periodically opening and closure of the cracks in the outer oxide layer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Since being identified in 1960s, hot corrosion has been given much attention and many achievements have been obtained.[1] Hot corrosion is initially caused by a sodium sulfate (Na2SO4) deposit resulting from the injection of salts into the engine and sulfur from the combustion fuel, so it remains an important property for large industrial gas turbines (IGTs) blade applications in those power plants located near to the seaside,[2] and where the fuel used for ITGs application is not of as high quality as for aero engines. Hot corrosion-resistant superalloys are extensively used in IGTs because of their outstanding mechanical strength and hot corrosion resistance. Due to the environment exacerbation and energy crises, more and more efficient power plants are required to reduce contamination and enhance thermal efficiency. One solution will be achieved by increasing the operating temperature of the turbines of the power plants.[3] Hence, the high-temperature strength of the hot corrosion-resistant superalloys must be further improved.
However, the requirements regarding high-temperature strength and hot corrosion resistance might come into conflict. For hot corrosion resistance, Cr is important since it can produce a protective Cr2O3 scale which is the most resistant oxide to Na2SO4-reduced hot corrosion.[4] Ti also plays an important role in resisting hot corrosion attack, as Ti is an effective element in gettering sulfur released by the salt and penetrating into the bulk.[5] Therefore, around 12 to 16 wt pct Cr along with approximately 3 wt pct Ti or more in the alloys are required for acceptable hot corrosion resistance.[5] However, Cr and Ti are very effective TCP forming elements but weak strengthening elements. High Cr and Ti limit the possibility to use other strengthening elements. In order to further improve the mechanical properties, one would have to make use of the design philosophy of typical aero engine materials with high refractory elements (Re, Mo, W, and Ta) contents and low Cr and Ti contents, while keep the hot corrosion resistance unaffected.[5,6] Therefore, refractory elements which are beneficial to hot corrosion resistance are of high importance. Mo and W are detrimental to hot corrosion resistance because of their acid oxides.[7,8] Our previous papers have revealed that Ta is a promising element for high strength hot corrosion-resistant superalloys. However, the improvement of Ta on hot corrosion is limited, so that the loss of hot corrosion resistance caused by Cr reduction cannot be compensated for by increasing the Ta content.[9,10] Recently, we have begun to study the effect of Re on hot corrosion resistance of Ni-based single crystal superalloys.
Re is the most effective solid strengthening element. Nevertheless, its influence on hot corrosion has rarely been reported. According to the few papers by Matsugi,[11,12,13] Caron and Czech,[14,15] hot corrosion resistance was improved remarkably by the addition of small amounts of Re. However, neither Re-containing products nor Re-enriched layer was observed by them or by others who have conducted hot corrosion tests of Re-containing superalloys.[16,17,18,19,20] Therefore, the effect of Re on hot corrosion behavior still needs further investigation.
Our previous paper has reported the beneficial effect of Re on hot corrosion and demonstrated the observation of preliminary morphologies.[21] In the present paper, detailed phase and microstructure analyses, and the mechanisms of the hot corrosion process in single crystal superalloys with and without Re addition will be discussed.
2 Materials and Experimental Procedures
The experimental methods have been described in our previous papers.[9,10]
2.1 Sample Preparation
The chemical compositions of the experimental alloys are listed in Table I (all data are given in wt pct).
Single crystal rods of the experimental alloys measuring 16 mm in diameter and 220 mm in length were fabricated by high rate solidification (HRS) method. All rods received a standard heat treatment. Hot corrosion specimens with dimensions of 20 mm × 10 mm × 1.5 mm were cut by electro-spark wire-electrode cutting from the as heat-treated rods, with the 20-mm-long sides of the samples kept parallel to the (001)-direction of the rods, while the other sides of the samples were in random orientations. The surfaces of the samples were prepared by grinding on the metallographic SiC paper to #800 and all the samples were chamfered before testing, in case the sharp corners bring any uncertainty or instability to the hot corrosion behavior. Ultrasonically cleaning with acetone and alcohol was performed after all the preparation processes. Scanning electron microscopy (SEM) equipped with an energy dispersive spectroscopy (EDS) was used to characterize the original microstructure of the sample.
2.2 Hot Corrosion Test and Sample Analysis
Hot corrosion testing was carried out at 900 °C. Specimens were placed on a hot Ni plate and sprayed with a saturated aqueous solution of Na2SO4 until their surfaces were coated with 0.3~0.5 mg/cm2 salt. The coated specimens were put into separate corundum crucibles and placed in static air at 1 atm at 900 °C to perform the hot corrosion test. The test was periodically interrupted every 20 hours and the samples were removed from the furnace for weight monitoring and salt-recoating. The time interval required for weight monitoring was 1 hour in every cycle. Each data point of the mass change was the average of three samples. Macroscopic observation and record were conducted in this time interval as well.
After a certain time, a sample will be removed from the test for microstructure analysis. X-ray diffraction (XRD), scanning electron microscopy (SEM) equipped with an energy dispersive spectroscopy (EDS), and electro-probe microanalyzer (EPMA) were used to identify the corrosion products and characterize the microstructure of the corroded samples. To prevent dissolving the water-soluble corrosion products in water, all the cross-sectioned samples were ground and polished in kerosene.[22]
3 Results
3.1 Hot Corrosion Kinetics and Macroscopic Morphologies
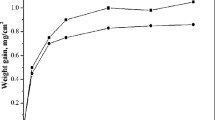
The hot corrosion kinetics and macroscopic morphologies of the experimental alloys have been discussed in our preliminary research paper.[21] In the present paper, the fitted curve was given (Figure 1). It can be seen that the E1 alloy followed a parabolic law with its incubation period being 100 hours. The overall corrosion kinetics of the Re-containing alloy followed a multistage parabolic law with the inflection points at 320 and 400 hours. The mass of E7 increased slowly during the whole test and the final mass gain was about 15 mg/cm2, this is much lower than that of alloy E1.

(a) Weight gain vs time curves for the single crystal superalloys E1 and E7 at 900 °C. (b) Multi-parabolic fitting curves of E7
The corresponding macroscopic morphologies showed that small protrusions appeared on the surfaces of E1 samples at 100 hours (Figure 2(b)), while the time that the protrusions appeared on E7 was 160 hours (Figures 2(e) and (f)). After 100 hours, the oxide scale on E1 became extremely fragile and started to crack and spall off. However, the E7 samples were nearly unchanged after the protrusions appeared. They were still compact and complete until 440 hours.

Macroscopic morphologies of the single crystal superalloys E1 and E7 corroded at 900 °C. (a) E1 for 20 h. (b) E1 for 100 h. (c) E1 for 200 h. (d) E7 for 20 h. (e) E7 for 100 h. (f) E7 for 160 h. (g) E7 for 440 h
3.2 XRD Results
The XRD results for the corrosion products are provided in Table II. For E1 alloy, as soon as the test started, a layer containing Cr2O3 and TiO2 formed. After 20 hours corrosion, TiO2 disappeared. Cr2O3 remained the main products on E1 until 100 hours. After that, NiO substituted the former Cr2O3 as the main corrosion product of the E1 sample and NiTiO3 became a constant product. Moreover, Na2SO4, which was detected before 100 hours, cannot be detected after that time. The composition of the products formed on E7 was more complex than that on E1. Before 120 hours, the main product on E7 was Cr2O3. In addition, several Ti-containing products TiO2, (Cr, Ti)TaO4 and Na2Cr2Ti6O16 existed. After 120 hours, NiTiO3 and NaTaO3 became the main products of E7. Weak peaks of NiO appeared after 220 hours. In addition, peaks of Na2SO4 can be detected throughout the whole test duration.
3.3 Surface and Cross Section Morphologies
According to the hot corrosion kinetics (Figure 1), the incubation period of E1 was 100 hours. The time when protrusions started to form on E1 was also 100 hours. Thus, morphologies of E1 sample at 100 hours are critical for understanding of the corrosion process and mechanism.
The surface and cross section morphologies of E1 at 100 hours are illustrated in Figures 3 and 4, in which the layers and products were identified according to the EDS and XRD analyses. Original surfaces of the sample were covered by a Na2SO4 film, sharp corners of the oxides below can be seen (Figure 3(b)). When comparing the cross section morphologies and compositions of protrusions on E1 and E7, we removed the residual salt on the samples by boiling water.

Surface morphologies of the corrosion scales formed on the E1 alloy after 100-h corrosion at 900 °C. The pictures were annotated according to the EDS and XRD analyses. (a) Macroscopic morphology of the protrusions on E1. (b) Higher magnification of the protrusion in (a). (c) Morphology of the protrusion after the sample was washed by boiling water. (d) Higher magnification of the morphology of the oxides on the surface

Cross section morphologies of the E1 alloy corroded for 100 h at 900 °C. The pictures were annotated according to the EDS and XRD analyses. (a) Macroscopic morphology of the cross section of E1 at 100 h. (b) Higher magnification of the protrusion in (a). (c) Cracks in the sub-surface. (d) Cross section morphology of the flat interface region in (a). (e) Transition region from protrusion to flat interface
At 100 hours, the surfaces of the E1 sample were covered by a dense NiTiO3 layer. Plenty of protrusions formed (Figures 3(a) and 4(a)). At the tip of the protrusion, the NiTiO3 layer spalled off (Figures 3(c) and 4(b)). The sub-surface layer was mixed oxides containing Al, Ni, Co, Cr, and Ta. Cracks parallel to and perpendicular to the surface of the protrusion can be seen (Figures 4(c) and (e)). Large amounts of inner sulfides CrSx formed, with those at the corrosion front almost continuous. In the flat surface region, the NiTiO3 layer and the inner oxide layer were complete and compact. Inner Al2O3 formed. The sizes of the CrSx particles were smaller and the quantity of them was much less than that of the protrusion region (Figure 4(d)).
As of 120 hours, a NiO-dominant layer formed on E1. Molten salt penetrated the outer NiO layer (Figures 5(a) and (e)). A dark dense salt layer can be seen at the middle of the oxide layer. Severe inner oxidation and sulfidation occurred. At 140 hours, a “hamburger” structure containing two fluxing layers and a NiO-dominant oxides layer formed (Figures 5(b) and (f)). Unlike 120 hours, the molten salt demonstrated the particle morphology with light gray oxides particles of Ni, Al, and Co dispersing in it. At the edge of the salt layer, a thin NiO layer existed (Figure 5(b)). Compared to 120 hours, amounts of inner oxides and sulfides increased rapidly. At the protrusion region, large amounts of holes formed (Figure 5(f)). As of 200 hours, the amount of the oxides particle dispersed in the molten salt increased (Figure 5(c)). Al2O3-dominant oxide network extended into the substrate. A thick porous NiO-dominant layer formed at the protrusion with the outermost part spalled off. The elemental distribution was illustrated in Figure 6.

Cross section morphologies of the E1 alloy after corrosion at 900 °C. The pictures were annotated according to the EDS and XRD analyses. (a), (b), and (c) Cross section morphologies of the flat interface region after 120-, 140-, and 200-h corrosion, respectively. (e), (f), and (g) Cross section morphologies of the protrusion after 120-, 140-, and 200-h corrosion, respectively. (d) and (h) Higher magnification of the regions “A” and “B” in (c)

Secondary electron micrograph and corresponding X-ray micrograph of the E1 alloy corroded for 180 h at 900 °C
For alloy E7, the protrusions formed at 160 hours. The outer NiTiO3 layer was not complete yet (Figures 7(a) and (b)). The amount of the inner oxides and sulfides was much less than that in alloy E1 at 100 hours. At 320 hours, when the mass change of the samples increased abruptly, the outer NiTiO3 layer became integral. The thickness of the corrosion as well as the amount of the inner corrosion products increased. No obvious spallation and cracks was observed in the NiTiO3 layer and the (Al, Co, Cr, Ta)-O oxides layer (Figures 7(c) and (d)). Holes can be seen in the (Al, Co, Cr, Ta)-O oxides layer. As of 440 hours, the samples were covered by a condensed Na2SO4 layer, with corners of oxides dispersing on it (Figures 8(a) and (b)). As the sample was washed by boiling water, the oxides below can be seen. The NiTiO3 layer on protrusions was still integral after 440 hours (Figure 8(c) through (e)). The amount of the inner sulfides was more than that at 320 hours. Cracks and holes can be seen. The NiTiO3 layer, (Al, Co, Cr, Ta)-O oxides layer, NaTaO3 layer and (Cr, Ti)-S sulfides layer were confirmed by EPMA analyses (Figure 9). In addition, a Re-enriched layer was observed in the cross section of E7 after 440-hour corrosion (Figure 9).

Cross section morphologies of the E7 alloy after corrosion at 900 °C. The pictures were annotated according to the EDS and XRD analyses. (a) and (b) Cross section morphologies of the flat interface region and the protrusion after 160-h corrosion, respectively. (c) and (d) Cross section morphologies of the flat interface region and the protrusion after 320-h corrosion, respectively

Surface and cross section morphologies of the E7 alloy corroded for 440 h at 900 °C. The pictures were annotated according to the EDS and XRD analyses. (a) Surface morphology of the E7 sample after hot corrosion test. (b) High magnification of surface morphology of the E7 sample. (c) Morphology of the protrusion after the sample was washed by boiling water. (d) Higher magnification of the morphology of the oxides on the surface. (e) and (f) The lateral views of the protrusion. (g) Cross section morphology of protrusion. (h) Cross section morphology of the flat surface region

Secondary electron micrograph and corresponding X-ray micrograph of the E7 alloy corroded for 440 hours at 900 °C. The scale consisted of an outer NiTiO3 layer, a sub-surface (Al, Co, Cr, Ta)-O oxides layer, an inner Al2O3 layer and an inner (Cr, Ti)-S sulfides layer. Besides, a Re-enriched layer formed
4 Discussion
4.1 Hot Corrosion Process
The melting point of pure Na2SO4 is 884 °C. As the test temperature approached 900 °C, the following reaction occurs in molten Na2SO4[23]:
The alloy will be oxidized by the molten salt and a three-layer structure containing an outer oxide layer, an inner oxide layer and an inner sulfide layer formed on the surface of the sample.[9] Due to the high Cr content and moderate Al content in E1 and E7, the initial corrosion layer was an outer Cr2O3-dominant layer,[24] an inner Al2O3 layer and an inner CrSx layer. As hot corrosion proceeded, the layer grew thicker, while the dissolution of the outer Cr2O3 by the oxide ions (O2−) in molten salt took place simultaneously.[1,25] The yellow-green morphologies of the samples surface (Figure 2) confirmed the formation of yellow Na2CrO4, which arose from the dissolution of Cr2O3 in Na2SO4:
4.1.1 E1
As the corrosion proceeded, Cr in alloy E1 was insufficient to maintain the completeness of the Cr2O3 scale. At 100 hours, oxide of Ni started to propagate, because large concentration of Ni enhanced its kinetic advantage. The Ni-containing oxides grew much more rapidly than Cr2O3.[26] Part of NiO reacted with the initially formed TiO2 to form NiTiO3. Consequently, a NiO-dominant layer formed on the E1 sample within a very short time (Figures 4 and 5).
Since NiO was very weak in resisting hot corrosion attack,[23] the released oxygen and sulfur traverse through the scale and caused inner oxidation and sulfidation. Due to the big PBR (Pilling-Bed-Worth Ratio) value of the inner corrosion productions (\( {\text{PBR}}_{{{\text{Al}}_{2} {\text{O}}_{3} }} \, = \,1.28,\,{\text{PBR}}_{{{\text{CrS}}_{x} }} \, = \,2.8\sim 3.87 \)), compressive stress was generated in the substrate. Simultaneous deformation of the scale and the substrate then occurred to accommodate the stress. As a result, protrusions were produced and tensile stress generated in the corrosion scale. Consequently, cracks formed at tip of the protrusion.
After protrusions and cracks were observed in E1 at 100 hours, the alloy experienced rapid corrosion because more corrodents were introduced in the substrate (Figures 5(a) and (e)). Figures 5 and 6 illustrated the subsequent fluxing of the NiO-dominant oxide layer, which exactly followed the Rapp-Goto theory.[26]
At 120 hours (Figure 5(a) and (e)), the penetration of the molten salt occurred but the fluxing did not start. The condensed salt was dark gray big blocks (Figure 5(a)). As the experiment went on, the O2− accumulated and began to react with the NiO-dominant oxides layer to form the corresponding ions at the NiO/Na2SO4 interface. As the ions diffused away from the NiO-dominant oxides layer, they decomposed into corresponding oxides particles and O2−. The re-precipitated oxides particles formed a thin layer at the outermost of the salt layer (Figures 5(b) and (f) and 6). As of 200 hours, the fluxing was considerable. In the reacting area, a large amount of re-precipitated oxides particles were dispersed in dark salt (Figure 5(d)). As the fluxing completed, the re-precipitated oxides formed a NiO-dominant thick loose and porous layer. Transformation and cracking of the scale were more remarkable at the fragile porous layer (Figure 5(g)).
With reference to Eq. [1], consumption of O2− by fluxing will cause increased activity of sulfur. Along with the fluxing of the outer oxide layer, severe inner sulfidation happened simultaneously (Figures 5(b) and (c) and 6). Due to the insufficient Cr content, liquid Ni-sulfides formed besides solid Cr-sulfides (Figures 5(c) and (h)). The liquid phase enhanced the diffusion rate of ions and accelerated the overall corrosion rate.
4.1.2 E7
Unlike E1, several Ti-containing products TiO2, (Cr, Ti)TaO4 and Na2Cr2Ti6O16 existed before 100 hours. At the beginning of the test, the scale was thin. Thus, the initially formed Cr2O3 and TiO2 can both be detected. As the corrosion proceeded, large amounts of Cr2O3 formed. Simultaneously, small amounts of TiO2 also formed and subsequently induced the formation of (Cr, Ti)TaO4 and Na2Cr2Ti6O16 by solid state reaction between TiO2 and corresponding oxides.
The integrated Cr2O3 layer was more resistant to the molten salt and more effective in inhibiting the inward diffusion of oxygen and sulfur. Thus, the formation of protrusions and penetration of salt on Re-containing alloy was delayed on E7.
Since the solubility of Ti in Cr2O3 is considerable, and the diffusivity of Ti was faster than Cr, TiO2 can form above the Cr2O3 layer.[27] Then, an additional integrate NiTiO3 layer formed above the Cr2O3 layer (Figures 7 and 8) by solid state reaction between TiO2 and NiO doped in Cr2O3:
(The calculation is carried out by HSC Chemistry® version 6.0, Outokumpu Research Oy, Finland). Cr2O3 as well as the spinel NiTiO3 are both effective in resisting Na2SO4-induced hot corrosion attack.
Because of the resistant oxide scale, most of the molten salt existed on the outer surface of the samples. Formation of the residual molten salt scale was another beneficial effect that promoted the better hot corrosion resistance of E7. According to Eq. [2], the dissolution rate of Cr2O3 was influenced by oxygen partial pressure (\( P_{{{\text{O}}_{2} }} \)). The salt scale lowered the \( P_{{{\text{O}}_{2} }} \) at the interface of oxides scale/salt and thus lowered the dissolution rate of Cr2O3. A virtuous cycle was established in E7. Therefore, surface morphologies of E7 samples were relatively stable during the whole period of the corrosion test (Figures 7 and 8).
4.2 Re Effect
According to the above hot corrosion test results, hot corrosion behavior of E1 and E7 was totally different. E7 followed multistage parabolic law, and the incubation period of E7 was much longer than that of E1. The microstructure of their corrosion scale was also different. The Cr2O3 on E7 lasted longer than on E1. More Ti-containing products on E7 were found than that on E1. NiTiO3 on E7 was comparatively more integral than that on E1. Large cracks formed and the outer oxide layer spalled off at the tips of the protrusions on the alloy E1. However, the outer layer of the tip was relatively integral on E7, which means no crack formed or cracks can be self-healing during hot corrosion.
Considering the hot corrosion behavior was closely related to the diffusion behavior, and the core factor affecting the diffusion behavior was the activity of the elements, it is believed that these distinguishing features in the E7 alloy were probably attributed to the increased activities of elements Cr and Ti caused by the Re addition.
The main factor responsible for longer incubation period of E7 was the higher stability of the Cr2O3 scale and the postponed formation of massive NiO. On the one hand, increased activity of Cr in E7 made the Cr2O3 scale on E7 stable for longer than that on E1, and enabled the self-healing ability of the Cr2O3 scale on E7. Thus, the formation of NiO was inhibited. On the other hand, increased activity of Ti promoted the formation of more TiO2 which was then sufficient to stabilize the non-protective NiO by forming protective spinel NiTiO3 (Eq. [3]). Finally, the Re-enriched layer can slow down the diffusion rate of Ni. This may also slow down the formation of massive NiO on E7 and stabilize the protective Cr2O3 scale during the corrosion test.
The multistage parabolic curve of E7 was probably because cracks in the outer scale open and close periodically. This phenomenon has also been observed in previous publications studying the oxidation behavior of Cr.[28,29] This observation can be also attributed to the increased activity of Cr in Re-containing alloy, which allows more Cr to diffuse outward to heal the cracks in the protective scale.
5 Conclusions
Hot corrosion behavior of Ni-based single crystal superalloys with 0 Re and 2 wt pct Re in molten Na2SO4 at 900 °C in static air was investigated using the “deposit recoat” method. The following conclusions can be drawn based upon analyses of the results.
-
1.
Re addition increased the stability of the Cr2O3 scale, facilitated the formation of TiO2, and promoted the formation of NiTiO3. Hot corrosion resistance was improved remarkably by Re addition.
-
2.
The overall corrosion kinetics of the Re-containing alloy followed a multistage parabolic law with the inflection points at 320 and 400 hours. It was probably because Re increased the activity of Cr and Ti, which resulted in the periodic opening and closing of the cracks in the outer oxide layer.
References
R.A. Rapp: Corros. Sci., 2002, vol. 44, pp. 209–221.
Y. Murata, R. Hashizurne, A. Yoshinari, N. Aoki and M.M. Fukni: Superalloys 2000, 2000, pp. 285-294.
S.Q. Zhao, X.S. Xie and G.D. Smith: Surf. Coat. Technol., 2004, vol. 185, pp. 178-183.
N. Otsuka and R.A. Rapp: J. Electrochem. Soc., 1990, vol. 137, pp. 53-60.
R. Burgel, J. Grossmann, O. Lusebrink, H. Mughrabi, F. Pyczak, R.F. Singer and A. Volek: Superalloys 2000, 2000, pp. 25-34.
R. C.Reed: The Superalloys Fundamentals and Applications, Cambridge University House, New York, 2006, pp. 157-162.
F.S. Pettit and G.H. Meier: Superalloys 1984, 1984, pp. 651-687.
K.R. Peters, D.P. Whittle and J. Stringer: Corros. Sci., 1976, vol. 16, pp. 791-804.
J.X. Chang, D. Wang, T. Liu, G. Zhang, L.H. Lou and J. Zhang: Corros. Sci., 2015, vol. 98, pp. 585-591.
J.X. Chang, D. Wang, G. Zhang, L.H. Lou and J. Zhang: Corros. Sci., 2017, vol. 117, pp. 35-42.
K. Matsugi, M. Kawakami, Y. Murata, M. Morinaga, N. Yukawa and T. Takayanagi: Tetsu to Hagane-Journal of the Iron and Steel Institute of Japan, 1992, vol. 78, pp. 821-28.
K. Matsugi, Y. Murata, M. Morinaga and N. Yukawa: Superalloys 1992, 1992, pp. 307-316.
M. Moniruzzaman, Y. Murata, M. Morinaga, R. Hashizume, A. Yoshinari and Y. Fukui: ISIJ Int., 2003, vol. 43, pp. 1244-1252.
N. Czech, F. Schmitz and W. Stamm: Surf. Coat. Technol., 1994, vol. 68, pp. 17-21.
P. Caron and T. Khan: Aerosp. Sci. Technol., 1999, vol. 3, pp. 513-523.
R.C. Reed, A. Sato, F. Karlsson and M. Hasselqvist: Superalloy 2012, 2012, pp. 197-204.
R. Hashizume, A. Yoshinari, T. Kiyono, Y. Murata and M. Morinaga: Superalloys 2004, 2004, pp. 53-62.
G.C. Fryburg, F.J. Kohl, C.A. Stearns and W.L. Fielder: J. Electrochem. Soc., 1982, vol. 129, pp. 571-585.
E. Liuy, Z. Zheng, X. Guan, J. Tong, L. Ning and Y. Yu: J. Mater. Sci. Technol., 2010, vol. 26, pp. 895-899.
G. Injeti, W. Alasdair, D. Hailiang, J. Burnell-gray and S. Datta: ECS Trans., 2008, vol. 6, pp. 105-115.
J.X. Chang, D. Wang, G. Zhang, L.H. Lou and J. Zhang: Superalloys 2016, 2016, pp. 177-185.
G.C. Fryburg, C.A. Stearns and F.J. Kohl: J. Electrochem. Soc., 1977, vol. 124, pp. 1147-1148.
J.A. Goebel and F.S. Pettit: Metall. Trans., 1970, vol. 1, pp. 1943-1954.
C.S. Giggins and F.S. Pettit: J. Electrochem. Soc., 1971, vol. 118, pp. 1782-1790.
D.K. Gupta and R.A. Rapp: J. Electrochem. Soc., 1980, vol. 127, pp. 2194-2202.
N. Birks, G.H. Meier and F.S. Pettit: Introduce to the high-temperature oxidation of metals, 2nd ed., Cambridge University Press, New York, 2006, pp. 115-119.
J. Litz, A. Rahmel, M. Schorr and J. Weiss: Oxid. Met., 1989, vol. 32, pp. 167-184.
P. Kofstad and K.P. Lillerud: J. Electrochem. Soc., 1980, vol. 127, pp. 2410-2419.
K.P. Lillerud and P. Kofstad: J. Electrochem. Soc., 1980, vol. 127, pp. 2397-2410.
Acknowledgments
This work was supported by the National Natural Science Foundation of China under Grant No. 51631008, the National Key Research and Development Program of China under Grant No. 2016YFB0701403, the National Training Programs of Innovation and Entrepreneurship for Undergraduates under Grant No. S201710705025 and the Xi’an Shiyou University “Materials Science and Engineering” Provincial Preponderant Discipline. The authors are grateful for these supports.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted March 13, 2018.
Rights and permissions
About this article

Cite this article
Chang, J.X., Wang, D., Liu, X.G. et al. Effect of Rhenium Addition on Hot Corrosion Resistance of Ni-Based Single Crystal Superalloys. Metall Mater Trans A 49, 4343–4352 (2018). https://doi.org/10.1007/s11661-018-4711-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-018-4711-3