
Abstract
Magnesium (Mg) alloys have been recently introduced as a biodegradable implant for orthopedic applications. However, their fast corrosion, low bioactivity, and mechanical integrity have limited their clinical applications. The main aim of this research was to improve such properties of the AZ91 Mg alloy through surface modifications. For this purpose, nanostructured fluoridated hydroxyapatite (FHA) was coated on AZ91 Mg alloy by micro-arc oxidation and electrophoretic deposition method. The coated alloy was characterized through scanning electron microscopy, transmission electron microscopy, X-ray diffraction, in vitro corrosion tests, mechanical tests, and cytocompatibility evaluation. The results confirmed the improvement of the corrosion resistance, in vitro bioactivity, mechanical integrity, and the cytocompatibility of the coated Mg alloy. Therefore, the nanostructured FHA coating can offer a promising way to improve the properties of the Mg alloy for orthopedic applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Metallic implants such as titanium, cobalt-chromium, and stainless steel have been widely used for fixation and repair of damaged bone.[1–3] Despite the active market of these traditional materials in the orthopedic industry, recently Mg alloys have attracted the attention of researchers due to their degradation capability to be used as orthopedic fixation plates and screw devices.[4–6] Biodegradability of Mg alloy implants can overcome the long-standing drawbacks associated with permanent implants, including foreign body response, delayed type hypersensitivity, and painful and expensive post-healing removal surgery.[7] However, there are serious barriers for clinical application of Mg alloys including fast corrosion rate, accompanied by hydrogen gas release, and limited bioactivity.[8–10] If the implants being made of Mg alloys are used to repair diseased bone tissue, they could possibly lose their mechanical properties before the healing of the bone tissue due to the rapid corrosion and low bioactivity.[1,11] Other concerns are about the cytocompatibility of Mg alloys implants and their ability for fast precipitation of osteoconductive minerals.[12,13]
Surface modification is a well-known strategy to enhance the corrosion resistance, interface biocompatibility, and bioactivity of metallic devices.[14] Coatings can protect the implants from corroding too quickly in vivo, and improve the interface bioactivity and biocompatibility.[15,16] More specifically, using bioactive ceramics as coating materials enhances the osseo-integration and bone healing procedure.[17]
Hydroxyapatite [HA: Ca10(PO4)6(OH)2] has been intensely studied over the years for medical applications due to its chemical similarity to the mineral phase of bone and tooth.[18] Incorporation of fluorine into the apatite structure to form fluoridated hydroxyapatite [FHA:Ca10(PO4)6OH2−x F x ; where x stands for the degree of fluoridation] will improve the physical and biological properties of hydroxyapatite.[19] Currently, hydroxyapatite partially substituted with fluorine has fascinated much interest and has been considered for clinical bone growth. Therefore, FHA can be a promising candidate for surface modification of metallic implants.
The bioactivity of a coating will be improved if it can be prepared in nanostructural configuration similar to the bone structure. Since the natural bone is composed of nanostructured hydroxyapatite, employing the bioceramic coating with nanostructured construct would more appropriately mimic the human bone. Thus, we synthesized the nanostructured FHA for our coating purposes in this study.
Electrophoretic deposition (EPD) is a convenient method to fabricate a great variety of ceramic coatings on metallic implants for biomedical applications.[20]
An appropriate approach to make a rough layer on Mg is anodization in high voltages, which is known as micro-arc oxidation (MAO).[20] MAO layers can be used to protect the substrate from corrosion and can act as an intermediate layer.[20]
To the best of our knowledge, there are no reports on the EPD of FHA on the micro-arc oxidized magnesium alloy. Thus, the main aim of our research was enhancing the corrosion resistance, bioactivity, mechanical stability, and cytocompatibility of Mg alloys by surface modification of an AZ91 Mg alloy employing a hybrid coating of MAO and EPD of nanostructured FHA.
2 Materials and Methods
2.1 Samples Preparation
Initial AZ91 magnesium alloy billet with the chemical composition presented in Table I was provided by Isfahan University of Technology. Plate samples (2 × 15 × 5 mm3) were machined from the AZ91 ingot and ground with SiC emery papers of up to 600 grits.
2.2 Preparation of FHA Powders
Preparation of FHA powder was performed according to the previous procedures.[21] The ball milling process was used for the preparation of the nanostructured FHA powder. The ball milling was conducted for 10 hours at rotational speed of 250 rpm and ball/powder ratio of 10/1.
2.3 MAO
The MAO process was performed using a DC power supply. An AZ91 sample and a stainless steel plate were adopted as the anode and the cathode of our setup, respectively. The electrolyte was composed of sodium hydroxide (200 g L−1) and sodium silicate (200 g L−1). The selection of electrolyte solution was based on the previous reports about MAO coating of magnesium alloys.[22–24] We used an alkaline (NaOH) solution containing sodium silicate (Na2SiO3) in order to form forsterite (Mg2SiO4) phase in the MAO matrix as this phase has a positive effect on enhancing the corrosion resistance and bioactivity of coating.[25,26] The concentrations of NaOH and Na2SiO3 were optimized based on the visual inspection of coating layers to have a uniform and thick coat on the surface. Samples were treated in the electrolyte for 0.5 hours with the cell potential increased gradually to 60 V.
2.4 Experimental SETUP for EPD Process
For EPD process, a suspension containing 100 g L−1 of FHA particles in methanol was prepared. The dispersion process was carried out using ultrsonication and magnetic stirring. The anode and the cathode were graphite and MAO samples, respectively. The EPD was performed for 3 min at 100 V.
2.5 Coatings Characterization
The phase composition of samples was characterized by X-ray diffraction (XRD, Philips X’Pert) and the grain size of the prepared FHA powders was calculated using the Williamson-Hall equation.[27] Scanning electron microscopy (SEM: Philips XL 30: Eindhoven) was used in order to analyze the surface of samples.
2.6 Electrochemical Test
An Ametek potentiostat (model PARSTAT 2273) was employed to perform the electrochemical corrosion tests. The samples were used as the working electrodes; a saturated calomel electrode (SCE) was used as the reference electrode and a platinum electrode was used as the counter electrode. As the corrosive medium, the simulated body fluid (SBF) was made according to the protocol described by Kokubo.[28] A scanning rate of 1 mVs−1 was applied during the polarization experiment, and the scan range of polarization test was in the scan direction of −250 to +500 mV versus open circuit potential. The perturbation voltage was 10 mV and EIS data were recorded from the frequency of 100 kHz to 10 mHz. Three samples were utilized for each experiment and the data were stated as mean ± standard deviation.
2.7 Immersion Test
The immersion tests were carried out in the SBF at 310 K (37 °C) for 0, 72, 168, 336, 504, and 672 hour. After that, the samples were dried and studied by SEM for bioactivity evaluation. Then, the samples were cleaned by chromic acid (180 g L−1 Cr2O3) to remove the corrosion products.[29]
The Mg ion release of samples was examined by inductively coupled plasma (ICP: PERKIN-ELMER 2380). To find the pH values of samples containing solution a pH-Meter (pH & ION meter GLP 22, Crison, Spain) was employed.
2.8 Compression Test
For compression test, the samples were prepared according to ASTM E9 standard. The compression test was carried out on an INSTRON 8562 testing machine at cross head displacement rate of 0.5 mm min−1 to measure the residual compressive properties of each sample.
2.9 Cytocompatibility Evaluation
A L-929 fibroblast cell line was employed for cytocompatibility examination. Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10 pct fetal bovine serum (Gibco), and 1 pct penicillin streptomycin was used as the cell culture media. The L-929 fibroblasts (3000 cells mL−1) were seeded in plates and incubated at 310 K (37 °C) in a humidified 5 pct CO2 atmosphere for 5 days. Three samples were used for each time point (2 days, 5 days, and 7 days) and cell viability tests were conducted at 310 K (37 °C). The DMEM medium was selected as a negative control. To perform MTT assay, 400 µL MTT (3-(4,5-dimethylthiazol)-2,5-diphenyltetrazoliumbromide) was added to wells and incubated at 310 K (37 °C) for 4 hour. Finally, the medium was replaced by 4 mL dimethylsulfoxide. A microplate reader (Hiperion MPR4+) was used to measure the absorbance of the samples. The cell viabilities were expressed as ODsample/ODnegative control × 100 pct, where ODsample and ODnegative control are the optical densities of the sample and the negative control, respectively. For cell attachment evaluation, cells were fixed on the sample by 2.5 pct glutaraldehyde solution and were dehydrated in 30, 50, 70, 90, 95, and 100 pct vol pct alcohol solutions. Statistical analysis was performed to evaluate the difference in cell viability by the analysis of variance. The statistical significance was defined as 0.05 and the data were presented in mean ± SD.
3 Results and Discussion
3.1 Coating Characterization
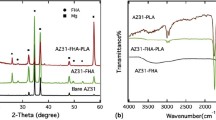
Figure 1 shows the SEM image of MAO layer (a), XRD pattern of MAO layer (b), TEM image of FHA particles (c), XRD pattern of FHA particles (d), SEM image of FHA coating (e), and XRD pattern of FHA coating.

SEM image of MAO layer (a), XRD pattern of MAO layer (b), TEM image of FHA particles (c), XRD pattern of FHA particles (d), SEM image of FHA coating (e), and XRD pattern of FHA coating
As can be seen in Figure 1(a), the surface of MAO sample is rough consisting of several pores. This configuration was formed by the molten oxide and gas bubbles released during micro-arcs. The MAO layer composed of one outer porous layer and one inner compact barrier layer. The inner layer, which was a metallurgical combination with the Mg substrate, is compact and uniform. The inner compact layer can protect the substrate from the corrosion. However, the outer layer of the MAO film could absorb more corrosive electrolyte and decrease the corrosion resistance of MAO layer on Mg.[30]
According to Figure 1(b), in the pattern of MAO coating, Mg, MgO, and Mg2SiO4 peaks could be found. MgO was formed by the dissolving Mg2+ from the substrate and the O2− from the electrolyte. At high temperature, both SiO2 and MgO are present and will form Mg2SiO4 (forsterite).[31] MgO and Mg2SiO4 were acting as the protective and bioactive phases, respectively in the MAO layer.[26,32]
A TEM image of the FHA nanoparticles is shown in Figure 1(c). According to this Fig., the size of particles is in the range of 50 to 100 nm and they present agglomerative morphologies with irregular shapes. The XRD results of the prepared FHA powder show expected crystallized peaks of FHA (Figure 1(d)). The grain size of FHA powder was obtained 22 nm according to Williamson–Hall equation.
The surface morphology of the FHA coating is shown in Figure 1(e). The surface of FHA coating is rough and porous with a netlike surface structure. It has been suggested that this structure can be beneficial for cell attachment and propagation, and may improve the connection of the implant to the surrounding bone tissue.[33]
According to Figure 1(f), for the FHA coating, besides the Mg, MgO, and Mg2SiO4 diffraction peaks, the diffraction peaks from the FHA particles were also detected by XRD, indicating that FHA has been coated as the main phase on the surface of MAO.
3.2 Electrochemical Test
Figure 2 and Table II present the results of polarization and EIS tests for the AZ91-, MAO-, and FHA-coated samples in the SBF. The values of corrosion current density (I corr), and corrosion potential (E corr) were derived from the polarization test (Figure 2(a); Table II). Based on conventional corrosion electrochemistry, the low corrosion current density and high corrosion potential are correlated to the good corrosion resistance.[34] According to Table II, it was found that the corrosion potential of coated samples is elevated slightly, while the corrosion current density is reduced significantly, as compared to the AZ91 samples. As shown in Table II, the order of the values of E corr (vs SCE) in different samples was as follows: FHA coating (−1.39 V) > MAO (−1.56 V) > AZ91 ( 1.6 V). The position of the I corr values were as follows: FHA coating (12.5 nA/cm2) < MAO (53,700 nA/cm2) < AZ91 (63,100 nA/cm2). Thus, the E corr values of the MAO- and FHA-coated sample are less negative than that of the AZ91 samples, and the I corr values for the MAO- and FHA-coated samples are lower as compared to the AZ91 samples, indicating that the MAO- and FHA-coated samples are less susceptible to corrosion. Furthermore, the E corr and I corr values indicate that the FHA-coated sample has a significantly higher corrosion resistance than those of the MAO samples.

Polarization (a), and EIS spectra of AZ91-, MAO-, and FHA-coated samples include the Bode (b) and phase (c) plots for these samples and an equivalent circuit which was proposed by ZSimpDemo 3.30d software to explain EIS spectra (d)
EIS spectra of AZ91-, MAO-, and FHA-coated samples were recorded and the Bode (b) and phase (c) plots for these samples are shown in Figure 2. Also Figure 2(d) shows an equivalent circuit which was proposed by ZSimDemo 3.30d software to explain EIS spectra. The EIS fitted results are presented in Table II. R p is used as a measure of coating resistance, where R t is used as a measure of polarization resistance which is the resistance at the coating/metal interface. The solution resistance (R s) between the reference and working electrodes mostly depends on the solution’s conductivity and cell geometry. R s is normally placed in series with the other elements in the equivalent circuit. C f and C dl represent the capacitance of the coating and electric double layer on the surface, respectively. C f indicates the diffusion of electrolyte solution owing to the chemical dissolution of coating, and C dl is attributed to the destruction of the coating. Moreover, R L (inductance resistance) and L (inductance) are associated with the low frequency inductance loop, which may cause by Cl ion adsorption.[35–39] It can be deduced from EIS spectra that R t and R p of AZ91 samples increased for MAO- and FHA-coated samples, suggesting that the MAO and FHA coating is more corrosion resistant than AZ91, which concurs with the results of polarization measurements. In Table II, Error (percent) shows the difference between the real data and the obtained results from the equivalent circuit model. The values of Error (percent) indicate that the real data have good conformity with the simulated data (Goodness of Fit).
3.3 In Vitro Bioactivity Evaluation
Figure 3 shows the SEM morphology of the AZ91- (a), MAO-, (b) and FHA-coated (c,d) samples after 672 hours immersion in the SBF. As shown in Figure 3(a), plenty of deep cracks were observed on the surface of uncoated AZ91 substrate. The cracks are probably along with the grain boundaries since they act as active regions for corrosion attacks. Moreover, the surface of MAO sample has been corroded and some corrosion defects such as corrosion pits and cracks can be seen representing that the MAO-coating layer has been corroded during the immersion process (Figure 3(b)). The existence of deep pits on the MAO-coating layer may be due to the electrolyte infiltration through the pores of coating. Figure 3(c) indicates that the lesser corrosion attacks have occurred on the surface and the FHA coating has maintained its shape stability. The cracks formation on the surface of magnesium alloy due to the presence of Cl− ion in a corrosive medium has been reported by other investigators.[38,40] Although MAO layer which composed of MgO and Mg2SiO4 is a protective layer against corrosion, due to its porous structure, the medium can diffuse through the pores and reach the AZ91 substrate leading to the formation of deep pits on the surface.[23,41] FHA coating can be considered as a bioactive ceramic layer.[21] Effective formation of bioactive minerals on its surface (bioactivity) as well as its corrosion resistance characteristics protects the substrate from the corrosion. The SEM images further identify that the surfaces of the FHA-coated samples were fully covered with the precipitates (Figure 3(c)) in leaf-like structures (Figure 3(d)). Comparing the corrosion and in vitro bioactivity behavior among the AZ91-, MAO-, and FHA-coated samples in different immersion times, we realized that the AZ91 sample has visually been more affected by corrosion compare to the MAO- and FHA-coated samples. On the other hand, the MAO- and FHA-coated samples have more uniform corrosion attack than the AZ91 sample, indicating the degree of corrosion damage was reduced for both of the MAO- and FHA-coated samples. Moreover, the density of precipitated white particles on the surface of FHA-coated samples (Figure 3(c)) is more than that of the MAO (Figure 3(b)) and AZ91 (Figure 3(a)) samples. When the samples were immersed into the SBF, FHA coating induced rapid precipitation of white particles on the surface. However, this phenomenon was not observed for the AZ91 sample. It is worth noting that the corrosion attack and precipitated white particles for the MAO sample is between that of the AZ91- and FHA-coated samples according to Figure 3(b). The leaf-like structure, observable in Figure 3(d), indicates the formation of bioactive minerals which is beneficial for the surface bioactivity. It has been reported that these precipitates have chemical composition close to the natural bone and can make an osteo-integrated interface between the implant and surrounding tissue.[42,43] According to the EDS analysis (Figure 3(e)), the precipitated white particles mainly consisted of Ca and P elements. Thus, the presence of Ca and P along with the observed leaf-like structure may confirm that the deposited particles contained of calcium phosphate compositions.[42,44]

(a through d) SEM morphology of the AZ91- (a), MAO-, (b) and FHA-coated (c,d) samples after 672 h immersion in the SBF. (e): EDS analysis from the precipitated layer on the surface of FHA-coated sample
3.4 Immersion Test
Recording the weight loss of the samples after immersion in the SBF solution shows that all samples displayed a rapid degradation after the first 72 hours, then the weight loss increased more slowly with immersion time, as indicated in Figure 4(a). In all time intervals, the weight loss of the AZ91 sample was higher than that of the MAO- and FHA-coated samples in the SBF solution, and the weight loss of the FHA coating was the lowest. The total weight loss of the AZ91 sample was approximately 67 mg after 672 hours of immersion, however, for the MAO- and FHA-coated samples, the total weight loss was 42 and 16 mg, respectively. The weight loss and weight gain of the Mg alloys are attributed to the degradation of Mg as well as the precipitation of apatite coating as the passive layer.[45] Both the degradation of Mg and apatite precipitation occurred at different rates for different samples due to their various surface-coating properties. The weight change of the samples in the SBF solution occurs as a result of the following two processes: (1) the degradation of Mg, and (2) the deposition of corrosion products on the surface. The degradation of Mg happened for all three groups of samples, but their degradation rates were notably different depending on the coating materials employed on the surface.[4,5] MAO and FHA coating can act as corrosion protective layers at the initial stage of immersion. However, the corrosion rate can enhance due to the formation of cracks on the surface at the initial stage of immersion. The corrosion of Mg alloys is mainly due to the presence of Cl− in the corrosive media.[40,46] Corrosion products may form continuously during the SBF soaking which can cause the weight gain.[45] Accumulation of corrosion products on the surface (Figure 3) protects the materials underneath and leads to a decrease in corrosion rate of the implant.

Weight loss (a), Mg ion concentration (b), and pH value (c) of the AZ91-, MAO-, and FHA-coated samples versus immersion time in the SBF and surface morphology of the AZ91- (d), MAO-, (e) and FHA-coated (f) samples immersed for 672 h in the SBF after cleaning the corrosion products
The corrosion behavior of biodegradable Mg alloys can also be studied by evaluation of Mg ion release concentration. ICP was used to determine Mg ionic concentrations in the SBF solution collected in our experiments (Figure 4(b)). The release on the first 72 hours was the highest, decreased (72 to 168 hours) to arrive at a steady state until the end of the immersion. The maximum Mg release was for the uncoated sample, showing the maximum corrosion rate. Both the MAO- and FHA-coated samples presented a significantly lower release of Mg ion. At first, the Mg ion concentration in the solution was enhanced due to the surface breakage (first 72 hours). Subsequently, the corrosion process slowed down because of the Mg phase-containing precipitates such as calcium magnesium phosphate or Mg-containing apatite resulted in consuming the Mg ion from the solution and reducing the Mg ion concentration (72 to 168 hours).[44,47] The Mg ion concentration reached a stable value during 168 to 672 hours by further increase in immersion time as in this stage we have both mild corrosion process and slow precipitation of the corrosion products on the surface.
Figure 4c shows the pH of the SBF solutions after immersion of the samples in them. Similar trend was observed in the pH value for all solutions. The pH value increased rapidly from 0 to 72 hours immersion, decreased slowly from 72 to 168 hours and reached a stable value afterward. The slower increase of the pH value of MAO and FHA sample-containing solutions during the first 72 hours can be due to the lower release of the OH− ion into the solution. Since at initial stage of immersion test, the corrosion attack is limited due to the presence of MAO- and FHA-coating layer on the surface which can prevent the AZ91 substrate from direct contact with corrosive media. During the 72 to 168 hours immersion, the pH value of all solutions decreased which is more pronounced in MAO and FHA samples. This can be due to the deposition of corrosion products including magnesium hydroxide and apatite.[45,48] Magnesium hydroxide is formed on the surface of immersed samples according to the below reaction (Reaction [1]) in which formation of magnesium hydroxide consumes the OH− from the SBF solution leading to a decrease in pH value[49]:
The biomineralization is initiated by electrostatic interaction of surface functional groups with the calcium and phosphate ions in the fluids. SBF is a supersaturated calcium phosphate solution. A chemical motivation of surface functional group can activate the nucleation of bioactive products. In this work, the existence of FHA as the coating material on the surface of magnesium alloy was considered as a nucleation template in the SBF incubation, offering many hydroxyl terminals, which are known to be efficient inducers of bioactive products nucleation. These hydroxyl terminals would attract Ca2+ and PO4 3− in the SBF and released Mg2+ from the substrate leading to the formation of apatite on the surface.[21,42,50] According to Figure 4, the pH decrease of the MAO and FHA sample-containing solutions is more rapid than that of the AZ91 sample due to the formation of bone-like apatite, which is in agreement with the outcome of the in vitro bioactivity evaluation (Figure 3).
Figure 4 also shows the surface morphology of the AZ91- (d), MAO- (e) and FHA-coated (f) samples immersed for 672 hours in the SBF after cleaning the products from the surface. The AZ91 sample indicates noticeable corrosion and the surface was full of web-like cracks and deep pits, resulting in a significant weight loss of AZ91 substrate, implying that the uncoated AZ91 alloy suffered from severe localized corrosion attack as shown in Figure 4(d). The local increase in pH, as well as the absence of any corrosion protective layer, will therefore cause the quick corrosion of the underneath plain surfaces. In addition, the presence of chloride ions in the SBF will damage the surface oxide film of Mg to induce random pits. Chloride ions facilitate the propagation of pits. Formation of pits is very harmful to the overall corrosion resistance and the damage of Mg alloy will advance by means of pitting corrosion.[51] Conversely, it could be seen that the MAO- (Figure 4(e)) and FHA-coated (Figure 4(f)) samples were subjected to lower corrosion attack compared to the uncoated AZ91 sample. After soaking in the SBF for 672 hours, the coated samples kept their shape integrities with the presence of a few pits on the surfaces. There existed only a few attacked spots on the as-cleaned MAO- and FHA-coated samples; and the depth of the corrosion pits was much shallower than that of the substrate. In other words, the residual area of the samples with the MAO and FHA coating was much larger than that of the substrate. The minimal damage was possibly due to the diffusion of water and electrolyte into the Mg substrate through the MAO and FHA coating pores, resulting in the corrosion attack.
3.5 Compression Test
The mechanical testing was carried out by examining the compression properties of the AZ91-, MAO-, and FHA-coated samples before and after immersion in the SBF for 4 weeks. Stress–strain curves for the AZ91-, MAO-, and FHA-coated samples are shown in Figure 5. According to Figure 5, the compressive strength of the FHA-coated AZ91 sample after 4 weeks immersion is more than that of the AZ91 sample after 4 weeks immersion. Therefore, the FHA-coated samples could better maintain its mechanical integrity, while the compressive strength of the uncoated AZ91 sample dropped significantly due to the corrosion and Mg ion release during the immersion. The compressive strengths of MAO- and FHA-coated samples remained at least 30 and 90 MPa higher than the uncoated AZ91 samples after 4 weeks of immersion, largely due to the slower corrosion rate which indicated that the FHA coating delayed the loss of the mechanical property of the substrate very effectively. In human natural bone, the compressive strength is reported to be in the range of 100 to 230 MPa for cortical bone and 2 to 12 MPa for cancellous bone,[52] and the results AQ also showed the compressive strength of FHA-coated samples after 4 weeks to be in the same range.

Stress–strain curves for the AZ91-, MAO-, and FHA-coated samples before and after 4 weeks immersion in the SBF
The mechanical integrity of an orthopedic implant should provide support to the fractured bone throughout the healing process, which may take several weeks. According to the results above, the FHA coating delayed the decrease of compressive properties of the AZ91 alloy to support the injured bone.
3.6 Cytocompatibility Evaluation
Figure 6 indicates the cell viability (pct of control) of L-929 cells after 2, 5, and 7 days of incubation on the uncoated and coated samples and SEM morphology of the cells cultured after 7 days incubation on the surfaces of the uncoated AZ91-(a), MAO-(b), and FHA-coated (c) samples.

Relative cell viability ( pct of control) of L-929 cells after 2, 5, and 7 days of incubation on the AZ91-, MAO-, and FHA-coated samples (a) and SEM morphology of the cells cultured for 7 days on the surfaces of the uncoated AZ91- (b), MAO-, (c) and FHA-coated (d) samples
According to Figure 6, the cell viability increased with time for coated samples. For the uncoated AZ91 samples, there was no significant increase in the cell viability in the whole incubation period. The cell viability on the uncoated AZ91 samples changed from 50 to 58 pct between 2 and 7 days of culture time (16 pct increase). The cell viability on the MAO-coated samples changes from 70 pct in 2 days of incubation to 85 pct in 7 days of incubation (21 pct increase) and for the FHA-coated samples changes from 160 pct in 2 days of incubation to 175 pct in 7 days of incubation (9 pct increase). Furthermore, FHA-coated samples show higher initial cell viability than that of the other samples, which indicates the superior cytocompatibility of FHA-coated samples.
Figure 6 also shows the morphology of cells, cultured for 7 days on the surfaces of AZ91-, MAO- and FHA-coated samples, which noticeably indicates the differences in the response of the cells to the different surfaces. For the uncoated AZ91 samples, only a few, less-spread, and minimal cell–cell interactions could be seen. Micro-cracks were also observed on the surface. For the MAO samples, a few cells attached on the surface. For the FHA-coated samples, the cells were confluent and the area occupied by the cells increased greatly on the surface. The cells spread and connected together with spherical granules on the cell surfaces, implying mineralization, and covered most of the sample so that it is hard to distinguish between the cells and the matrix. Therefore, better cell adhesion and spreading can be accomplished on the FHA-coated samples than MAO and AZ91 samples. Also, in comparison with the AZ91 sample, more cells had spread and attached to the surface of MAO.
Degradation of Mg is accompanied by the releasing hydrogen gas and formation of corrosion product layer. The formation of corrosion products and hydrogen gas prevents the proper cell attachment. This can explain why MAO- and FHA-coated samples have better cell attachment compared to the AZ91 substrate. Also the presence of the Ca2+ ion which is an essential element for the cell signaling causes better absorption of favor proteins such as fibronectin and vitronectin, which can promote cell attachment.[53] Cell attachment examination is the first step in the cell–material surface interactions study, which in turn can influence subsequent relations between materials and surrounding tissues.[54]
The substrate (AZ91 magnesium alloy) and the coating material (FHA) were selected among the well-known cost-effective materials for biomedical applications. Moreover, the coating technique (MAO/EPD) is industrially efficient as an easy and inexpensive technique. However, before commercializing this product, long-term in vivo animal examinations for confirming the results, as well as the clinical trial tests are necessary.
4 Conclusions
AZ91 Mg alloy was coated with nanostructured FHA by MAO and EPD method. Detailed characterizations of the coated alloy indicated that the corrosion resistance, the in vitro bioactivity, the mechanical stability and the cytocompatibility of the Mg alloy were improved after the surface modification. Therefore, the nanostructured FHA can make an effective coating to improve the properties of the Mg alloy as a biodegradable bone implant for orthopedic applications.
References
MP Staiger, AM Pietak, J Huadmai and G Dias, Biomaterials 2006, vol. 27, pp. 1728-1734.
E. Salahinejad, M.J. Hadianfard, D.D. Macdonald, M. Mozafari, D. Vashaee, and L. Tayebi, Mater. Lett. 2013, vol. 97, p. 162–65.
E Salahinejad, MJ Hadianfard, DD MacDonald, S Sharifi-Asl, M Mozafari, KJ Walker, AT Rad, SV Madihally and L Tayebi, PloS one 2013, vol. 8, p. e61633.
YF Zheng, XN Gu and F Witte, Materials Science and Engineering: R: Reports 2014, vol. 77, pp. 1-34.
M Razavi, M Fathi, O Savabi and M Boroni, Research and Reviews in Materials Science and Chemistry 2012, vol. 1, pp. 15-58.
M Razavi, M Fathi, O Savabi, SM Razavi, BH Beni, D Vashaee and L Tayebi, Materials Letters 2013, vol. 113, pp. 174-178.
F Witte, Acta Biomaterialia 2010, vol. 6, pp. 1680-1692.
F Witte, V Kaese, H Haferkamp, E Switzer, A Meyer-Lindenberg, CJ Wirth and H Windhagen, Biomaterials 2005, vol. 26, pp. 3557-3563.
HM Wong, KWK Yeung, KO Lam, V Tam, PK Chu, KDK Luk and K Cheung, Biomaterials 2010, vol. 31, pp. 2084-2096.
M Razavi, M Fathi, O Savabi, BH Beni, D Vashaee and L Tayebi, Colloids and Surfaces B: Biointerfaces 2014, vol. 117, pp. 432-440.
M Razavi, MH Fathi, O Savabi, D Vashaee and L Tayebi, Physical Science International Journal 2014, vol. 4, pp. 708-722.
Y Xin, T Hu and PK Chu, Acta biomaterialia 2011, vol. 7, pp. 1452-1459.
M. Razavi, M. Fathi, O. Savabi, D. Vashaee, and L. Tayebi: Ann. Biomed. Eng., 2014, vol. 42, pp. 2537–50.
M. Yazdimamaghani, M. Razavi, D. Vashaee, and L. Tayebi: Surf. Eng., 2014, vol. 30, pp. 920−26.
AR Boccaccini, S Keim, R Ma, Y Li and I Zhitomirsky, Journal of The Royal Society Interface 2010, vol. 7, pp. S581-S613.
M. Razavi, M. Fathi, O. Savabi, D. Vashaee, and L. Tayebi: J. Biomed. Mater. Res. Part A, 2014, DOI:10.1002/jbm.a.35324.
H-M Kim, Current opinion in solid state and materials science 2003, vol. 7, pp. 289-299.
L.L. Hench and J. Wilson: An Introduction to Bioceramics, World Scientific, Singapore, 1993.
H-W Kim, Young-Min Kong, Chang-Jun Bae, Yoon-Jung Noh and Hyoun-Ee Kim, Biomaterials 2004, vol. 25, pp. 2919-2926.
H Hornberger, S Virtanen and AR Boccaccini, Acta biomaterialia 2012, vol. 8, pp. 2442-2455.
MH Fathi and E Mohammadi Zahrani, Journal of Crystal Growth 2009, vol. 311, pp. 1392-1403.
HF Guo, MZ An, HB Huo, S Xu and LJ Wu, Applied surface science 2006, vol. 252, pp. 7911-7916.
W Shang, B Chen, X Shi, Y Chen and X Xiao, Journal of Alloys and Compounds 2009, vol. 474, pp. 541-545.
YM Wang, JW Guo, ZK Shao, JP Zhuang, MS Jin, CJ Wu, DQ Wei and Y Zhou, Surface and Coatings Technology 2013, vol. 219, pp. 8-14.
F Chen, H Zhou, B Yao, Z Qin and Q Zhang, Surface and Coatings Technology 2007, vol. 201, pp. 4905-4908.
M Kharaziha and MH Fathi, Ceramics International 2009, vol. 35, pp. 2449-2454.
GK Williamson and WH Hall, Acta Metallurgica 1953, vol. 1, pp. 22-31.
T Kokubo and H Takadama, Biomaterials 2006, vol. 27, pp. 2907-2915.
KY Chiu, MH Wong, FT Cheng and HC Man, Surface and Coatings Technology 2007, vol. 202, pp. 590-598.
X Lin, L Tan, Q Zhang, K Yang, Z Hu, J Qiu and Y Cai, Acta biomaterialia 2013, vol. 9, pp. 8631-8642.
Y Zhang and C Yan, Surface and Coatings Technology 2006, vol. 201, pp. 2381-2386.
H-L Wu, Y-L Cheng, L-L Li, Z-H Chen, H-M Wang and Z Zhang, Applied Surface Science 2007, vol. 253, pp. 9387-9394.
Y.K. Lee, K.M. Kim, and K.N. Kim, Surf. Coat. Technol., vol. 195, pp. 252–57.
X Cui, Y Li, Q Li, G Jin, M Ding and F Wang, Materials chemistry and physics 2008, vol. 111, pp. 503-507.
R.C. Alkire, D.M. Kolb, J. Lipkowski, and P. Ross: Advances in Electrochemical Science and Engineering, Wiley, New York 2006.
J Degner, F Singer, L Cordero, AR Boccaccini and S Virtanen, Applied Surface Science 2013, vol. 282, pp. 264-270.
ASTM Standard G105-89: ASTM Standards, Philadelphia, 2004.
Y Zhang, C Yan, F Wang and W Li, Corrosion Science 2005, vol. 47, pp. 2816-2831.
AM Fekry, AA Ghoneim and MA Ameer, Surface and Coatings Technology 2014, vol. 238, pp. 126-132.
G Song, A Atrens, X Wu and B Zhang, Corrosion Science 1998, vol. 40, pp. 1769-1791.
L Zhao, C Cui, Q Wang and S Bu, Corrosion Science 2010, vol. 52, pp. 2228-2234.
JR Jones, Acta biomaterialia 2013, vol. 9, pp. 4457-4486.
A Shahini, M Yazdimamaghani, KJ Walker, MA Eastman, H Hatami-Marbini, BJ Smith, JL Ricci, SV Madihally, D Vashaee and L Tayebi, International journal of nanomedicine 2014, vol. 9, pp. 167-181.
M Razavi, MH Fathi and M Meratian, Materials Letters 2010, vol. 64, pp. 2487-2490.
M Yazdimamaghani, M Razavi, D Vashaee and L Tayebi, Materials Letters 2014, vol. 132, pp. 106-110.
M. Razavi, M. Fathi, O. Savabi, S.M. Razavi, F. Heidari, M. Manshaei, D. Vashaee, and L. Tayebi: Appl. Surf. Sci., 2014, vol. 313, pp. 60–66.
M Razavi, MH Fathi and M Meratian, Materials Characterization 2010, vol. 61, pp. 1363-1370.
M Razavi, M Fathi, O Savabi, D Vashaee and L Tayebi, Materials Science and Engineering: C 2014, vol. 41, pp. 168-177.
NT Kirkland, N Birbilis and MP Staiger, Acta biomaterialia 2012, vol. 8, pp. 925-936.
M. Mozafari, M. Mehraien, D. Vashaee, and L. Tayebi: Nanocomposites-New Trends and Developments, InTech, 2012.
R Hahn, JG Brunner, J Kunze, P Schmuki and S Virtanen, Electrochemistry Communications 2008, vol. 10, pp. 288-292.
M Razavi, MH Fathi and M Meratian, Materials Science and Engineering: A 2010, vol. 527, pp. 6938-6944.
K Cai, A Rechtenbach, J Hao, J Bossert and KD Jandt, Biomaterials 2005, vol. 26, pp. 5960-5971.
TV Kumari, U Vasudev, A Kumar and B Menon, Trends in Biomaterials and Artificial Organs 2002, vol. 15, pp. 37-41.
Acknowledgments
The authors are thankful for the contributions of Isfahan University of Technology, Torabinejad Dental Research Center, Oklahoma Center for Advancement of Science and Technology (Grant no. AR131-054 8161), AFOSR (Grant no. FA9550-10-1-0010) and the National Science Foundation (NSF, Grant no. 0933763).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Manuscript submitted May 17, 2014.
Rights and permissions
About this article

Cite this article
Razavi, M., Fathi, M., Savabi, O. et al. In Vitro Analysis of Electrophoretic Deposited Fluoridated Hydroxyapatite Coating on Micro-arc Oxidized AZ91 Magnesium Alloy for Biomaterials Applications. Metall Mater Trans A 46, 1394–1404 (2015). https://doi.org/10.1007/s11661-014-2694-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-014-2694-2