
Abstract
The selective oxidation of a CMnSi transformation-induced plasticity (TRIP) steel during intercritical annealing (IA) in a N2 + 10 pct H2 gas atmosphere with a dew point (DP) in the range from 213 K to 278 K (−60 °C to +5 °C) was investigated by transmission electron microscopy. The decarburization during IA resulted in a fully ferritic matrix at the TRIP steel surface. Annealing in high DP gas atmospheres resulted in a reduction of the oxide layer thickness at the surface and an increase of the depth of the subsurface internal oxidation. The experimental results were compared to the calculations of the DP for the transition from internal to external oxidation based on the Wagner model. The evolution of the surface oxide composition during annealing was analyzed thermodynamically by means of the chemical potential diagram for the surface oxides. In the high DP atmosphere conditions, mainly, Mn-rich xMnO·SiO2 (1 < x < 2) oxides were formed at the surface, while Si-rich xMnO·SiO2 (x < 1) oxides were formed by internal oxidation. The use of a high DP gas atmosphere is therefore advantageous to induce internal selective oxidation and reduce the amount of surface oxides. It also leads to the formation of Mn-rich xMnO·SiO2 (1 < x < 2) oxides.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The interaction of the surface of steel with the gas atmosphere in the annealing furnace of a hot-dip galvanizing (HDG) line results in the reduction of iron oxide. The alloying elements, Mn, Si, Al, Ti and V, are subject to oxidation. This is due to the fact that the N2 + 5 to 10 pct H2 gas mixture contains water vapor. The H2O dissociation reaction H2O ↔ H2 + 1/2 O2 determines the oxygen partial pressure (\( P_{{{\text{O}}_{ 2} }} \)), which is, albeit extremely low, still oxidizing toward Mn, Si, and Al. The amount of water vapor in the annealing furnace is represented by the dew point (DP) temperature. The furnace gas atmosphere typically has a low DP, resulting in the external selective oxidation of Mn and Si. Selective oxidation occurs when a solute atom is oxidized rather than the matrix element. The selective oxidation phenomenon occurring at the surface (external oxidation) and in the subsurface (internal oxidation) of most industrial steel grades has been investigated in detail for interstitial free steel, dual phase steel, and transformation-induced plasticity (TRIP) steel.[1–7] The presence of film-forming surface oxides, in particular amorphous a - xMnO·SiO2 (x < 0.9) and a -SiO2 oxides, leads to a deterioration of the wettability of the intercritically annealed strip by the molten Zn during the galvanizing process.[8–10] These oxides also act as barriers to the interdiffusion of Fe and Zn during galvannealing process.[11–14] Annealing in a high DP gas atmosphere has been shown to result in internal oxidation rather than external oxidation, and the control of the DP has been proposed as a method to improve the quality of Zn and Zn-alloy coatings on Si alloyed steels.[4–6]
Several theories for internal oxidation have been proposed. Wagner[15] proposed a theory for the internal oxidation of binary alloys. The original theory was based on the assumption of a single oxidizing element forming a single oxide with a zero solubility product. Numerous improvements of the original model were proposed. Böhm and Kahlweit[16] and Rapp[17] extended the model to account for a none-zero oxide solubility product. A model for multi-component alloys was proposed by Niu and Gesmundo.[18] Huin et al.[19] developed an internal oxidation model for multi-component ferrous alloys assuming a finite solubility product. Brunac et al.[20] then extended the model proposed by Huin et al.[19] to account for the possibilities that a large fraction of precipitates may act as diffusion barriers and that some precipitates may form and re-dissolve at a later stage due to the formation of other precipitates.
Despite the obvious usefulness of these models for the description of selective internal oxidation, they are still unable to capture the complex non-equilibrium external and internal oxidation processes in steel, in particular the morphology, the composition and the spatial distribution of the oxides.
The present study focuses on the thermodynamic aspects of the influence of the DP on the selective oxidation of CMnSi TRIP steel during intercritical annealing (IA) in gas atmosphere conditions used in industrial HDG line annealing furnaces. A comprehensive analysis of the oxides was done by field emission-transmission electron microscopy (FE-TEM) of cross-sectional samples prepared by the focused ion beam (FIB) technique. A comparison between the experimental results and calculated results based on the Wagner theory and a computational thermodynamic model was also made. Finally, the influence of the selective oxidation and decarburization on the subsurface microstructure was investigated by TEM.
2 Internal–External Selective Oxidation Transition
Wagner[15] proposed a theory for the internal oxidation of binary A-B alloys which allows for the isothermal internal oxidation of the alloying element B, forming a single oxide BO ν . The molar concentration of B, \( N_{\text{B,crit}}^{{ ( {\text{O)}}}} \), at which transition from internal to external oxidation occurs is given by Eq. [1]:
Here, g* refers to the critical BO ν oxide volume fraction when the transition from internal to external oxidation occurs, \( N_{\text{B}}^{{ ( {\text{O)}}}} \) is the bulk molar concentration of B, \( N_{\text{B}}^{{ ( {\text{S)}}}} \) is the oxygen molar concentration at the surface, D O is the diffusion coefficient of oxygen, D B is the diffusion coefficient of B, V m is the molar volume of the alloy, and \( V_{{{\text{BO}}_{v} }} \) is the molar volume of the oxides. The theory predicts that oxidation of B is always external when \( N_{\text{B}}^{{ ( {\text{o)}}}} > N_{\text{B,crit}}^{{ ( {\text{O)}}}} \).
When the flux of oxygen is significantly less than that of B, external oxides will grow laterally on the surface, and form a continuous layer. This layer blocks the inward diffusion of oxygen. Wagner stated that when the volume fraction of the oxide, g, reached a critical value g*, the transition from internal to external oxidation should occur. Rapp[21] tested Wagner’s theory for Ag-In alloys and found that the transition occurred when g* = 0.3. Equation [1] also provides a useful means to understand internal oxidation, as it predicts that when the alloy is exposed to conditions where \( N_{\text{B}}^{{ ( {\text{S)}}}} \) is large, e.g., high DP atmosphere, the value of \( N_{\text{B,crit}}^{{ ( {\text{O)}}}} \) increases, and the transition from internal to external oxidation should take place at higher concentrations of B. An increase of the effective D B due, for example, to a reduction of the grain size, results in a decrease of \( N_{\text{B,crit}}^{{ ( {\text{O)}}}} \). A temperature increase for a given gas atmosphere also influences the transition from external to internal oxidation as both the oxygen permeability, i.e., the product \( N_{\text{B}}^{{ ( {\text{S)}}}} \cdot D_{\text{O}} \), and the corresponding product for the alloying element B, \( N_{\text{B}}^{{ ( {\text{S)}}}} \cdot D_{\text{B}} \), increase at higher temperature.
3 Experimental
Industrially cold-rolled full-hard CMnSi TRIP steel containing 2.2 mass pct Mn and 1.4 mass pct Si was used for the present work. The Mn/Si mass pct ratio of the CMnSi TRIP steel was 1.57 and bare spots were not present in the Zn coating surface after the galvanizing simulation.[22] The dimensions of the sheet panels used for the annealing experiments were 220 mm in length, 120 mm in width, and 1.0 mm in thickness. The samples were intercritically annealed in an IWATANI SURTEC HDG simulator manufactured by IWATANI Inc. This HDG simulator consists of a main unit, a gas mixing station, a hybrid humidification system, and a control system. The infra-red furnace in the main unit was used for the IA simulation. The process gasses, N2, H2 and He, were mixed in the gas mixing station and the gas mixture was humidified in the hybrid humidification system. The control system allowed for the precise control of DP level and the composition of the gas atmosphere.
The IA was carried out in a N2 + 10 pct H2 gas atmosphere with DPs of 213 K, 243 K, 263 K, 273 K, and 278 K (−60 °C, −30 °C, −10 °C, 0 °C, and +5 °C). The DPs of 213 K and 243 K (−60 °C and −30 °C) were chosen as typical low DP atmospheres. These low DP conditions are close to those used in the annealing furnace in industrial continuous annealing lines (CAL) and HDG lines. The DPs of 273 K and 278 K (0 °C and +5 °C), for which internal selective oxidation of Mn and Si occurs, were chosen as high DP atmospheres. Huin et al.[19] provided a formula to calculate \( P_{{{\text{O}}_{ 2} }} \) and mass concentration of dissolved oxygen at the steel surface (\( C_{\text{O}}^{{ ( {\text{S)}}}} \)) in an annealing atmosphere containing water vapor. The saturation vapor pressure of H2O (\( P_{{{\text{satH}}_{ 2} {\text{O}}}} \)), in atm, is obtained from the DP temperature, in °C, by means of the following equation:
The corresponding oxygen partial pressure, \( P_{{{\text{O}}_{ 2} }} \), in atm, is given by the following equation:
Here, T is the temperature in K and \( P_{{{\text{H}}_{ 2} }} \) is partial pressure of hydrogen in atm. In the present study, a 10 pct H2 gas atmosphere was used with balance N2. The total pressure was 1 atm. The value of \( P_{{{\text{H}}_{ 2} }} \) was therefore taken to have the constant value of 0.1 atm. \( C_{\text{O}}^{{ ( {\text{S)}}}} \), in ppm, was then calculated using the following equation:
The calculated oxygen partial pressure at each of the DP conditions used in this work is listed in Table I.
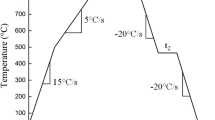
Prior to the continuous annealing simulations, small coupons of 15 × 15 mm2 were taken from larger panel samples and their surface was mirror-polished with a 1 μm diamond suspension in order to allow for precise cross-sectional TEM observations of the surface without interferences of the roughness of the strip surface. The annealing cycle consisted of a heating stage to the IA temperature of 1093 K (820 °C) at a heating rate of +3 K/s, a holding stage at the IA temperature for 47 s and a rapid cooling stage. The samples were also rapidly cooled to room temperature by N2 gas after: (a) heating to 923 K (650 °C), (b) heating to the IA temperature, and (c) holding at the IA (Figure 1). The cooling rates achieved with the N2 gas were in the range of −20 K/s to −30 K/s.

Schematic of the thermal cycle used for the experiments
The microstructures of the annealed samples were investigated by means of FE-TEM. The cross-sectional TEM samples were prepared by the FIB technique in a FEI Quanta 3D FEG. The TEM samples were analyzed in a JEOL JEM-2100F FE-TEM operated at 200 keV. The oxide composition was determined by means of energy dispersive spectroscopy (EDS) using a 1 nm size electron beam. The Oxford INCA software was used for the quantitative analysis of the composition. The reported oxide compositions are based on ten separate EDS measurements carried out on the oxides in the TEM samples.
The comprehensive experimental observations of the selective oxidation of Mn and Si were compared to the calculated critical transition temperatures for internal to external oxidation in the CMnSi TRIP steel using Eq. [1]. For the calculation of Eq. [1], the values of \( N_{\text{O}}^{{ ( {\text{S)}}}} \) in the gas atmosphere for the DP in the range from 213 K to 278 K (−60 °C to +5 °C) were adjusted from the values of \( C_{\text{O}}^{{ ( {\text{S)}}}} \) obtained using Eqs. [2], [3], and [4]. The parameters and equations used for the calculation are listed in Table II.[21,23–25] The critical value for g*, for the transition from internal to external oxidation occurs, was set equal to 0.3, as suggested by Rapp.[21] D O, D Mn, and D Si are the ferrite diffusion coefficients in cm2/s of oxygen, Mn, and Si, respectively. V MnO, \( V_{{{\text{SiO}}_{ 2} }} \) , and V Fe are the molar volumes of SiO2, MnO and ferrite, in cm3/mol, respectively. It was assumed that the matrix was fully ferritic at the surface and in the subsurface during IA. This assumption is reasonable as the surface of TRIP steel is fully decarburized when exposed to high temperature in the annealing gas atmosphere.[6,22,26] \( N_{\text{Mn}}^{{ ( {\text{O)}}}} \) and \( N_{\text{Si}}^{{ ( {\text{O)}}}} \), the Mn and Si molar concentrations in the steel were given by the nominal steel composition. Because all values needed for the calculation of Eq. [1] are known except for T, the critical temperatures (T*) for the transition from internal to external oxidation can be calculated.
A model proposed by Huin et al.[19] was also used to predict the internal oxidation of a steel containing 2.2 mass pct Mn and 1.4 mass pct Si during an isothermal IA for 47 seconds in a gas atmosphere with a low DP of 243 K (−30 °C) and with a high DP of 278 K (+5 °C). The calculation was carried out assuming that only two oxides, MnO and SiO2 could precipitate. The equations for the diffusion coefficients and the solubility products listed in Table II were used for the calculation. K MnO and \( K_{{{\text{SiO}}_{ 2} }} \) are the solubility products in (ppm)2 and (ppm)3 of MnO and SiO2, respectively. The theoretical model and the method to calculate the thermodynamic equilibrium are summarized in Appendix. The DP of 243 K (−30 °C) was selected as a low DP because the value of the lowest DP of 213 K (−60 °C) was too low to lead to the oxidation of the dissolved Mn at the surface to MnO in the initial step of the numerical calculation. This is due to the fact that the product of \( C_{\text{O}}^{{ ( {\text{S)}}}} \) and Mn mass concentration is less than the value of the solubility product of MnO. This initial condition prevents all Mn oxidation in the numerical calculation.
Additional equilibrium thermochemical calculations to obtain chemical potential diagrams were carried out using the Fact-Sage (version 6.4) thermochemical software and database.
Decarburization of the steel surface during IA was also studied by means of dilatometry and TEM observation of the samples prepared by the FIB technique. Dilatometry was used to measure the austenite volume fraction in the bulk alloy. Dilatometry specimens with dimensions of 10 mm in length, 3.5 mm in width, and 3 mm in thickness were machined with their length parallel to the transverse direction of sheets. Dilatometry experiments were carried out in a Bahr 805 pushrod dilatometer in a vacuum better than 3 × 10−4 mbar. The heat treatment cycle consisted of heating to 1093 K (820 °C) with a heating rate of +3 K/s, holding at the IA temperature of 1093 K (820 °C) for 47 seconds, further heating to full austenitization temperature of 1373 K (1100 °C) at a heating rate of +3 K/s, and a final slow cooling to room temperature.
4 Results and Discussion
4.1 TRIP Steel Surface Decarburization
The selective oxidation behavior of TRIP steel has been assumed to take place for a microstructure containing austenite and ferrite by some authors,[4,5,27] while others[19,20] developed a model assuming a fully ferritic surface. The presence of austenite at the steel surface during IA of a TRIP steel is critical to the oxidation mechanism. Gong et al.[5] predicted that kinetics of oxidation should be significantly influenced by the presence of austenite at the IA temperature, as the diffusivity of Mn is very much lower than that of Si in austenite. In addition, the solubility of Mn in the austenite matrix is higher than that of Si. Both factors should result in a limited Mn oxidation. The austenite fraction at the surface of a TRIP steel during IA is not known as the surface gradually decarburizes in the annealing atmosphere. The presence of austenite at the surface during IA was therefore studied in order to precisely predict the oxidation kinetics.
In the bulk system, the austenite volume fraction can easily be measured by dilatometry. Figure 2 shows the change of the austenite volume fraction in the TRIP steel during heating to 1093 K (820 °C) with a heating rate of +3 K/s, holding at 1093 K (820 °C) for 47 seconds, and heating to 1373 K (1100 °C). The austenite volume fraction was calculated using the lever rule, as illustrated in Figure 2(a). The microstructure of the bulk sample of the TRIP steel contained 25 vol. pct austenite directly after heating to the IA temperature and 45 vol. pct austenite after 47 seconds at the IA temperature.

Dilatometry results of a CMnSi TRIP steel during heating to 1093 K (820 °C), holding 47 s at 1093 K (820 °C) for and heating to 1373 K (1100 °C). (a) Dilatation of the TRIP steel during the heat treatment. The austenite volume fraction, f γ, was calculated using the lever rule, as illustrated in the inset. (b) Temperature dependence of the austenite volume fraction
Although the austenite fraction in the subsurface is difficult to measure precisely, the presence of austenite can be tested by the direct observation of the subsurface microstructures. Figure 3 shows the transition of the subsurface microstructure during annealing in low DP [213 K (−60 °C)] conditions. After heating to 923 K (650 °C), carbides were embedded in the recrystallizing ferrite grains located close to the surface, as shown in the EDS elemental maps of Figure 3(b). Carbides containing some Mn were clearly detected by EDS. After heating to 1093 K (820 °C), ferrite grain growth occurred. A small grain of martensite, surrounded by ferrite grains containing dislocation, was observed at a depth of approximately 1 μm below the surface (Figures 3(c) and (d)). After the IA at 1093 K (820 °C) for 47 seconds, there was no martensite or retained austenite island in the subsurface within 1 μm below the surface (Figure 3(e)).

Cross-sectional TEM micrographs showing the transition of the subsurface microstructure during annealing in low DP [213 K (−60 °C)] conditions. α and α′ are ferrite and martensite, respectively. (a) STEM bright-field image of the subsurface microstructure after heating to 923 K (650 °C). (b) EDS elemental maps for Mn, C, and O of the area corresponding to A in (a). (c) STEM bright-field image of the subsurface microstructure after heating to 1093 K (820 °C). (d) Enlarged area corresponding to B in (c). (e) STEM bright-field image of the subsurface microstructure after IA at 1093 K (820 °C) for 47 s
Figure 4 shows the transition of the subsurface microstructure during annealing in high DP [278 K (+5 °C)] conditions. After heating to 923 K (650 °C), the carbide particles containing Mn were also observed close to the surface, as shown in the EDS data of Figure 4(b). After heating to 1093 K (820 °C), grain growth occurred and only ferrite grains were present in the subsurface. Martensite grains were observed at a depth of 2 µm below the surface. Mn enrichment of one of the martensite grains, clearly shown by the EDS analysis in Figure 4(d), points to Mn partitioning to intercritical austenite. After an IA at 1093 K (820 °C) for 47 seconds, no martensite island was observed in the subsurface. A single martensite island was observed 3 µm below the surface. The depth of the decarburization after IA in low and high DP conditions is therefore at least 3 µm. These observations indicate that no austenite was present in the subsurface during IA as a result of the decarburization and the formation of Mn containing oxides. Slightly more martensite islands were observed in the subsurface after annealing in 278 K (+5 °C) DP conditions but the subsurface was free of martensite within 2 µm below the surface.

Cross-sectional TEM micrographs showing the transition of the subsurface microstructure during annealing in high DP [278 K (+5 °C)] conditions. α and α′ are ferrite and martensite, respectively. (a) STEM bright-field image of the subsurface microstructure after heating to 923 K (650 °C). (b) EDS elemental maps for Mn, C, and O of the area corresponding to A in (a). (c) STEM bright-field image of the subsurface microstructure after heating to 1093 K (820 °C). (d) Enlarged area corresponding to B in (c). (e) STEM bright-field image of the subsurface microstructure after IA at 1093 K (820 °C) for 47 s. (f) TEM image of the martensite in the area C of (e)
The decarburization was not severe for temperatures below 923 K (650 °C) because carbides were still observed near the surface after heating to 923 K (650 °C). After heating to 1093 K (820 °C), no evidence for the formation of intercritical austenite, i.e., observation of martensite or retained austenite at room temperature, was found close to the surface. Instead, martensite was present at a depth greater than 2 μm below the surface. After an IA for 47 seconds, martensite islands were only observed at much greater depths below the surface. Previous studies[6,22,26] have shown that retained austenite or martensite was not observed at the surface or in the subsurface of TRIP steel after continuous annealing simulations. The absence of retained austenite at the surface after the annealing clearly revealed that the surface and a 1 to 2 µm thick subsurface layer are fully decarburized and that the formation of oxides of the alloying elements occurs in a ferrite matrix near the surface. SEM observation of the microstructure of the subsurface after annealing of a TRIP steel was also done by Liu et al.[28] They reported that the surface of a TRIP steel was decarburized during annealing in a N2 + 5 pct H2 gas atmosphere with a DP of 283 K (+10 °C) and that a 20-μm thick ferrite layer was formed near the strip surface. According to the numerical simulation of internal oxidation of a Mn- and Si-added steel by Huin et al.,[19] the oxidation depth and the reaction layer during annealing cycle should be less than 1.5 μm. It can therefore be concluded that there is no austenite at the surface and in the subsurface layer approximately 2 μm below the surface during IA of TRIP steel. The selective oxidation of Mn and Si will therefore occur in a fully ferritic microstructure.
4.2 Selective Oxidation in High and Low DP Gas Atmospheres
The oxides species formed after IA of CMnSi TRIP steel are expected to be MnO, SiO2, MnO·SiO2, and 2MnO·SiO2.[29] However, the Mn/Si atomic ratio of the oxides does often not conform to the composition of these well-known simple or compound oxides.[5,22] In the present study, the compound oxides were described by means of the notation xMnO·SiO2, where x is the Mn/Si atomic ratio. When x = 0, the oxide is SiO2. The values x = 1 and x = 2 correspond to the compounds MnSiO3 and Mn2SiO4, respectively. The xMnO·SiO2 oxides are also classified into two groups depending on the value of x: Mn-rich oxides have x > 1, and Si-rich oxides have x < 1.
Figure 5 shows cross-sectional TEM observations of the evolution of the surface oxidation during annealing in low DP [213 K (−60 °C)] conditions. After heating to 923 K (650 °C) (Figure 5(a)), a continuous 5 nm thin layer of xMnO·SiO2 oxides formed at the surface. There where the grain boundaries intersect the surface, the oxide layers were found to be slightly thicker. In the subsurface, recrystallization resulted in a small ferrite grain size because the steel had been heavily deformed during cold rolling. As the temperature was increased to 1093 K (820 °C) (Figure 5(b)), ferrite grain growth and decarburization occurred in the subsurface. Si-rich oxide regions and thicker Mn-rich oxides regions developed separately at the surface. Further IA for 47 seconds led to the thickening of these layers (Figure 5(c)). While the thin oxide films consisted mainly of Si oxide, the Mn content was higher in the thicker oxide layer.

Cross-sectional TEM micrographs for CMnSi TRIP steel after annealing in low DP [213 K (−60 °C)] conditions. STEM bright-field images of the surface after (a) heating to 923 K (650 °C), (b) heating to 1093 K (820 °C), and (c) after IA at 1093 K (820 °C) for 47 s
Figure 6 shows cross-sectional TEM observations of the evolution of the surface oxidation during annealing in high DP [278 K (+5 °C)] conditions. After heating to 923 K (650 °C) (Figure 6(a)), a thin layer of external oxides formed at the surface and recrystallization took place in the subsurface. A 5 nm thin layer of xMnO·SiO2 oxides and MnO were present in the grain boundaries. During heating to 1093 K (820 °C) (Figure 6(b)), external and internal oxidation of Mn and Si occurred simultaneously. In the subsurface region, fine ferrite grains developed within a network of Mn and Si oxides. Si and Mn were internally oxidized at the boundaries between recrystallizing grains, thereby preventing their growth. This type microstructure with fine subsurface grain boundaries pinned by internal oxides was also observed by Gong et al.[5] During IA (Figure 6(c)), the depth of the internal oxidation extended to 1 μm, whereas the thickness of the external oxide layers did not increase appreciably. External xMnO·SiO2 oxide clusters were present on all the locations where the grain boundaries intersected the surface.

Cross-sectional TEM micrographs for CMnSi TRIP steel after annealing in high DP [278 K (+5 °C)] conditions. STEM bright-field image of the surface after (a) heating to 923 K (650 °C), (b) heating to 1093 K (820 °C), and (c) after IA at 1093 K (820 °C) for 47 s
4.3 External to Internal Oxidation Transition Due to DP Control
Figure 7 shows cross-sectional TEM micrographs used to detect the transition from internal to external oxidation by variation of the DP and the direct observation of the location of the oxidation front. The samples were annealed at the IA temperature in a gas atmosphere with a DP of 213 K, 243 K, 263 K, 273 K, and 278 K (−60 °C, −30 °C, −10 °C, 0 °C, and +5 °C) prior to quenching. In the low DP [213 K and 243 K (−60 °C and −30 °C)] conditions, external selective oxidation occurred exclusively. There was a clear transition from external to internal oxidation at a DP of 263 K (−10 °C). The value of \( P_{{{\text{O}}_{ 2} }} \) at the IA temperature in this DP condition was 7.38 × 10−22 atm. Separate regions of external and internal oxidation developed. A thinner oxide layer formed at the surface when the internal oxidation was favored. The depth of internal oxidation was 400 to 600 nm. In high DP [278 K (+5 °C)] conditions, the oxidation front extended 1 to 1.2 μm below the surface. Annealing in the 278 K (+5 °C) DP gas atmosphere resulted in a thinner layer of external oxidation and a larger depth of internal oxidation for Mn and Si.

Cross-sectional TEM micrographs of CMnSi TRIP steel after IA in a N2 + 10 pct H2 gas atmosphere with increasing DP: (a) 213 K (−60 °C), (b) 243 K (−30 °C), (c) 263 K (−10 °C), (d) 273 K (0 °C), and (e) 278 K (+5 °C). The transition from external oxidation to internal oxidation can be seen to occur at a DP of 263 K (−10 °C)
Figure 8 presents the oxidation behavior for 2.2 pct Mn and 1.4 pct Si added steel as a function of gas atmosphere DP and temperature. Figure 8(a) shows the critical temperature, T*, where the transition from internal to external oxidation of MnO and SiO2 is predicted to take place. The values of T* were calculated using Eq. [1]. The Wagner theory only considers binary alloys. T* was therefore calculated for MnO (\( T_{\text{MnO}}^{*} \)) in a 2.2 mass pct Mn Fe-Mn steel and for SiO2 (\( T_{{{\text{SiO}}_{ 2} }}^{*} \)) in a 1.4 mass pct Si Fe-Si steel. The calculations indicate that for a given atmosphere, external oxidation should take place above T* while internal oxidation should occur below T*. For the case of multiple alloying systems such as the CMnSi TRIP steel used in the present work, application of the model is more complicated. If internal oxidation of Mn and Si occurs concurrently, the transition from internal to external oxidation will occur at temperatures lower than \( T_{\text{MnO}}^{*} \) and \( T_{{{\text{SiO}}_{ 2} }}^{*} \) which were calculated for binary alloys. This is due to the fact that the accumulated external oxides will block the inward diffusion of oxygen, i.e., when two types of oxides form, the blocking effect will be enhanced. The surface oxygen will also be consumed for the oxidation of two elements, Mn and Si. Hence, the value of \( N_{\text{O}}^{{ ( {\text{S)}}}} \) will decrease due to the competing oxidation of Mn and Si. The fact that a less reactive alloying element also acts as oxygen getter is known as “secondary gettering.”[30] Mataigne et al.[31] have proposed an alternative model for the selective oxidation of multi-component alloys. They proposed an equation which includes the additive effect of the alloying elements:

DP and temperature effects on the oxidation behavior of the steel containing 2.2 mass pct Mn and 1.4 mass pct Si. (a) T* for MnO and SiO2 in different DP gas atmospheres. The T* values were calculated using Eq. [1]. (b) T* for MnO + SiO2 in different DP gas atmospheres. The T* values were determined by assuming the additive effect of MnO and SiO2 and calculated by means of Eq. [5]
The values of T*, calculated using Eq. [5], i.e., assuming the additive effect of MnO and SiO2, are indicated in Figure 8(b). The new T* values for the CMnSi TRIP steel decreased when the additive effect was considered. The T* in an actual microstructure will also decrease due to the grain boundary contribution to the effective D B. The influence of grain boundaries on the transition of internal/external oxidation was studied by Wang[32] and Liu et al.[33]This grain boundary effect may be large in the present case since the material undergoes recrystallization during annealing. Other factors affecting Wagner’s criterion for the external to internal oxidation transition are the solubility of the oxides and the compound oxides, MnO·SiO2 and 2MnO·SiO2. Driving a new criterion from this better basis would however require much additional theoretical work. Several attempts have been made to include these factors and extend the transition criterion of the Wagner theory,[18–20,34] but these models require data which can only be obtained from specific experiments.
The general trend that internal oxidation is favorable in a high DP gas atmosphere is in agreement with the Wagner model. However, the calculated T*s did not correspond to that of the actual T*, 1093 K in 263 K (820 °C in −10 °C) DP conditions. Both the Wagner and Mataigne criteria predict that, in all DP conditions, only external oxidation should occur because the IA temperature is always above the transition boundary, which is indicated by the dashed lines in Figures 8(a) and (b). Experimentally, external oxidation occurred for IA in 213 K and 243 K (−60 °C and −30 °C) DP conditions. Both external and internal oxidation occurred during IA in 263 K, 273 K, and 278 K (−10 °C, 0 °C, and +5 °C) DP conditions (Figure 7). The transition occurred in the 263 K (−10 °C) DP conditions. In the present study, the value of g* was set to 0.3. To exactly match the calculated transition DP with the experimentally observed transition DP of 263 K (−10 °C), the value of g* should be 2.4, which exceeds the maximum possible volume fraction of 1. The discrepancy was very likely due to some of the assumptions that have been used in the calculation, in particular the fact that the solubility products for the oxides were assumed to be zero and the fact that the formation of compound oxides was neglected.
Another discrepancy was that, in the 278 K (+5 °C) DP conditions, external oxidation occurred mostly during heating to 923 K (650 °C) and internal oxidation was dominant during annealing at 1093 K (820 °C), although external oxidation should be preferred at higher temperature. This phenomenon can be explained if the kinetics of the oxidation is considered. If the steel had been annealed at 923 K (650 °C) for a longer time, internal oxidation may have occurred after external oxidation. More explicitly, in 278 K (+5 °C) DP conditions, the oxidation occurred in the following sequence: temporary external oxidation during heating to 923 K (650 °C), combined external and internal oxidation during heating to 1093 K (820 °C) and exclusive internal oxidation during IA. It should be pointed out that although steel is annealed in conditions where internal oxidation is the preferred oxidation mode, the formation of some amount of external oxides is inevitable since the oxidation always starts from the surface.
Figure 9 shows calculated concentration profiles of a steel containing 2.2 mass pct Mn and 1.4 mass pct Si. Huin’s model was used for the calculations.[19] The calculation was carried out assuming that only two oxides, MnO and SiO2, could precipitate and their solubility products are finite values. After isothermal IA for 47 seconds in 243 K (−30 °C) DP conditions, enrichment in Mn occurs only at the surface, whereas enrichment in Si occurs over a non-negligible distance below the surface, (Figures 9(a) and (c)). The total fraction of Si was found to reach a minimum value at a depth of 0.04 μm. The Si oxidation front depth was 0.2 μm. This indicates that most of the Mn and Si were externally oxidized and only small fraction of Si was internally oxidized. After isothermal IA in 278 K (+5 °C) DP conditions, the total fraction of Mn and Si was found to reach a minimum at a depth of 0.06 and 0.08 μm, respectively (Figure 9(b)). The Mn and Si oxidation front depth extended to 0.04 and 0.25 μm, respectively, (Figure 9(d)). In both DP conditions, the Si oxidation front depth was larger than that of Mn. This is due to the lower solubility product of SiO2. Both Mn and Si can be externally oxidized due to a relatively large oxygen content at the outermost surface. However, the concentration of oxygen in the subsurface is much smaller than on the outer surface. In this condition, Si will always be oxidized first because Mn, whose oxide has larger solubility product, cannot form an oxide with small amounts of oxygen. Some important results shown by the calculations were in agreement with the experimental results. The calculations show that annealing in higher DP conditions results in smaller amounts of surface oxides and larger depths of internal oxidation. However, the calculated oxidation front depth was still too small, compared to the depth measured experimentally; for example, in 278 K (+5 °C) DP conditions, the calculated oxidation front depth is 0.25 μm (Figure 9(d)), whereas the depth measured experimentally was approximately 1.2 μm (Figure 7).

Numerical simulation of the internal oxidation of a steel containing 2.2 mass pct Mn and 1.4 mass pct Si.[19] An isothermal IA for 47 s was simulated. (a) Mn and Si mass concentration profiles for 243 K (−30 °C) DP conditions. (b) Mn and Si mass concentration profiles for 278 K (+5 °C) DP conditions. (c) MnO and SiO2 mass concentration profiles for 243 K (−30 °C) DP conditions. (d) MnO and SiO2 mass concentration profiles for 243 K (+5 °C) DP conditions
4.4 Surface Oxide Composition
Oxide species formed on the TRIP steel surface were found to have a composition which changed depending on the DP conditions. Figure 10 shows the distribution of the Mn/Si atomic ratio of the oxides formed during an IA in different DP conditions. For each DP condition, ten EDS measurements randomly taken on the oxides are indicated in Figure 10. Both Si-rich and Mn-rich oxides were present on the surface after an IA in 213 K and 243 K (−60 °C and −30 °C) DP conditions. Smaller amounts of the Si-rich oxides were present after an IA in 263 K (−10 °C) DP conditions. After annealing in higher DP [273 K and 278 K (0 °C and +5 °C)] conditions, no Si-rich oxide was formed and only xMnO·SiO2 (1 < x < 2) oxides were present on the surface. The Si-rich oxides were only present as internal oxides in the subsurface.

Distribution of the Mn/Si atomic ratio of the oxides formed during an IA in a N2 + 10 pct H2 gas atmosphere with a DP of 213 K, 243 K, 263 K, 273 K, and 278 K (−60 °C, −30 °C, −10 °C, 0 °C, and +5 °C). The Mn/Si ratio was calculated from EDS data
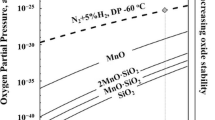
One of the limitations of the original Wagner theory is that it does not consider the often complex structure and composition of the oxides. Suzuki et al.[29] suggested an alternative model to predict the stability of the oxide species using the thermodynamic calculation of the chemical potential of the Fe-Mn-Si-O system. They used a chemical potential diagram to determine the stability of the oxide species in specific conditions. A similar approach of the interpretation was used in the present study. Figure 11 shows the calculated equilibrium mole fraction of the oxide species at 1093 K (820 °C) for each DP condition. The calculation indicates that, at the given IA temperature [1093 K (820 °C)], SiO2 and MnO·SiO2 are thermodynamically stable in 213 K (−60 °C) DP conditions, while the Mn-richer 2MnO·SiO2 becomes more stable at higher oxygen partial pressures, i.e., at a DP of 278 K (+5 °C). This calculation accounts for the absence of the Si-rich surface oxide at the steel surface after annealed in high DP conditions. The tendency that the fraction of Mn rich oxide increases at higher DP conditions is in very good agreement with the experimental results. Two types of the surface oxides, Mn-rich and Si-rich oxides, were formed separately during an IA in 213 K and 243 K (−60 °C and −30 °C) DP conditions, whereas only one type of oxide, xMnO·SiO2 (1 < x < 2), was formed on the surface during an IA in 273 K and 278 K (0 °C and +5 °C) DP conditions (Figure 10). It is also reasonable that Si-rich oxides were not formed at the surface but in the subsurface because the local oxygen partial pressure is much lower in the subsurface than at the outermost surface. Si-rich oxide is more stable than Mn-rich oxide at lower oxygen partial pressure.

Calculated equilibrium mole fractions of the Mn-Si compound oxides which are predicted to be formed on a TRIP steel containing 0.1 mass pct C, 2.2 mass pct Mn, and 1.4 mass pct Si at 1093 K (820 °C) in a N2 + 10 pct H2 gas atmosphere with a DP of 213 K, 243 K, 263 K, 273 K, and 278 K (−60 °C, −30 °C, −10 °C, 0 °C, and +5 °C)
Non-stoichiometry was found for the O/(Mn + Si) atomic ratio of the oxides at the surface. Figure 12 clearly shows that the surface oxides were non-stoichiometric, while the internal oxides were stoichiometric. If the surface oxides were stoichiometric compounds, the O/(Mn + Si) atomic ratio should be 1, 4/3, 3/2, and 2 for MnO, 2MnO·SiO2, MnO·SiO2, and SiO2, respectively. However, the O/(Mn + Si) atomic ratio for the surface oxides was always less than one, whereas the ratio for the internal oxide was mostly in the range from 1 to 2. This suggests that the reaction at the surface may be different from in the subsurface and that the surface oxides had either a metal ion excess or an oxygen deficiency. Töpfer and Dieckmann[35] reported that the deviation from stoichiometry, δ of Mn2SiO4+δ was dependent on the oxygen activity. They found that a more highly oxidizing atmosphere increased the value of δ, indicating an increased metal ion deficit in the compound. In contrast, a reducing atmosphere results in the creation of oxygen vacancies. Burn and Neirman[36] pointed out that oxygen vacancies could be easily created by the loss of oxygen from the crystal lattice in low oxygen partial pressure conditions at high temperature, according to the following reaction:

Distribution of the O/(Mn+Si) atomic ratio of the external and internal oxides formed during an IA in a N2 + 10 pct H2 gas atmosphere with a DP of 213 K, 243 K, 263 K, 273 K, and 278 K (−60 °C, −30 °C, −10 °C, 0 °C, and +5 °C). The O/(Mn+Si) ratio was calculated from EDS data
Here, the evaporation of an oxygen ion at the surface yields one oxygen vacancy in the oxide, \( V_{\rm{O}}^{ \cdot \cdot } \). The local evaporation of oxygen in a gas atmosphere of low oxygen partial pressure is a possible reason for the oxygen deficiency of the surface oxide. It is at present unclear how the presence of oxygen vacancies may affect the selective oxidation.
The Wagner theory[15] and its extensions[16,17,19,20] view the selective oxidation process as the internal precipitation of an oxide which takes place when the product of the concentrations of oxygen and the solute atom in the matrix exceeds the solubility limit of the oxide. Also, it is assumed that the kinetics of the internal oxidation is mainly controlled by diffusion of the oxygen and the solutes in the matrix. These basic assumptions are quite reasonable when calculating the kinetics of oxidation. However, non-stoichiometry of the surface oxide compounds implies that the actual phenomena occurring during surface oxidation may be quite different from the oxide precipitation during internal oxidation. It is known that the surface oxides are grown by the transport of ions, electrons, and/or holes through the oxide layer. Migration of ions and electrons through non-stoichiometric compounds is one special characteristic of the surface oxidation which does not apply to internal oxidation.
Figure 13 shows the chemical potential diagrams for a 0.1 pct C-Mn-Si steel at 1093 K (820 °C) in 213 K and 278 K (−60 °C and +5 °C) DP conditions. These diagrams provide useful information to predict the reaction paths for the oxide precipitation during an isothermal heat treatment. The reaction paths, indicated by the dashed lines in Figure 13, represent the local steel composition at the surface due to the reduction of the solute content by oxide formation. The Mn/Si ratio of the solutes is determined by the stoichiometry of the oxide species which are formed, e.g., two Mn and one Si atoms are consumed simultaneously during the precipitation of 2MnO·SiO2. The reaction path in the diagram for low DP conditions (Figure 13(a)) suggests that MnO·SiO2, a primary oxide, and SiO2, a secondary oxide, will be formed simultaneously during an isothermal heat treatment. This is consistent with the experimental observation that when the IA temperature was reached, two types of surface oxides were formed: Mn-rich and Si-rich oxides (Figure 5). These oxides grew separately during the IA holding stage. The reaction path in the diagram for the high DP conditions (Figure 13(b)) suggests that as oxidation progresses, the following sequence of oxide species is formed: (1) 2MnO·SiO2 → (2) 2MnO·SiO2 + MnO·SiO2 → (3) MnO·SiO2 → (4) MnO·SiO2 + SiO2. In practice, the formation of SiO2 as a surface oxide is almost fully prevented because internal oxidation starts to take place before the final stage of the oxidation sequence is reached. In addition, because the solubility product of SiO2 is much smaller than that of MnO, Si tends to be oxidized internally, as predicted by the calculation in Figure 9(b). These interpretations are also in agreement with the experimental observation that the Mn/Si atomic ratios for the surface oxides formed during an IA were between 2 and 1, i.e., 2MnO·SiO2 and MnO·SiO2, and that no Si-rich oxide was observed at the surface (Figure 10).

Oxidation phase diagrams for 0.1 pct C-Mn-Si steel at 1093 K (820 °C) in (a) low DP 213 K (−60 °C) and (b) high DP 278 K (+5 °C) conditions
It is now well established that the use of a high DP is advantageous for inducing internal oxidation and forming fewer surface oxides during IA of Si-containing TRIP steel. Another advantage, according to the present study, is that, in high DP conditions, Mn-rich oxides are thermodynamically more stable than Si-rich oxides and thus Mn-rich oxides are more likely to form. As these Mn-rich oxides do not form oxide films, they are less harmful to galvanizability than the film-forming Si-rich oxides. There are two reasons for this. First, the MnO reduction by Al in the Zn bath is thermodynamically possible, and the thermodynamic driving force for the alumino-thermic reaction is higher than that for SiO2. Second, Mn oxides form a particle morphology rather than a film morphology. Therefore, although the formation of oxide particles is inevitable during the continuous annealing, they can easily be detached and still allow for the wetting of the steel surface by the liquid Zn. The chemical potential diagram for the surface oxides can be used to determine the oxidation potential and alloy content for which the formation of Si-rich oxides can be avoided.
5 Conclusions
The selective oxidation of a CMnSi TRIP steel during IA in a N2 + 10 pct H2 gas atmosphere with a DP in the range from 213 K (60 °C) to 278 K (+5 °C) was investigated by FE-TEM. The decarburization and selective oxidation during IA resulted in a fully ferritic matrix at the TRIP steel surface and in the subsurface region ~2 μm below the surface. No evidence for the formation of intercritical austenite, i.e., room temperature martensite or retained austenite, was observed at the surface. Instead, martensite islands were present at least 2 to 3 μm below the surface after IA. The influence of the presence of austenite on the oxidation kinetics is therefore very limited.
External oxidation occurred exclusively during IA in low DP gas atmospheres. Annealing in higher DP conditions resulted in a thinner layer of external oxidation and a greater depth of internal oxidation. The transition of external to internal oxidation was observed in a gas atmosphere with a DP of 263 K (−10 °C). Even though the surface oxides were still present at the surface, the thickness of the surface oxides was effectively reduced by increasing the DP to activate internal selective oxidation. The experimental observation was generally in agreement with the prediction of the transition from internal to external oxidation by Wagner model, but the experimentally observed transition conditions [DP of 263 K (−10 °C)] were not equal to the calculated transition conditions..
The oxide species formed on the surface during IA were consistent with the primary oxide predicted by the chemical potential diagram. In low DP annealing conditions MnO·SiO2 formed as a primary oxide, while SiO2 formed as a secondary oxide during an isothermal heat treatment. In the high DP conditions, Mn-rich xMnO·SiO2 (1 < x < 2) oxides were mainly formed before internal Si-rich xMnO·SiO2 (x < 1) oxides could form. The formation of the Si-rich xMnO·SiO2 (x < 1) oxides was therefore suppressed at the surface in high DP annealing conditions. The use of high DP is clearly advantageous not only to induce internal selective oxidation and form less surface oxides but also in order to form Mn-rich oxides rather than Si-rich oxides.
References
J. Mahieu, S. Claessens, B.C. De Cooman: Metall. Mater. Trans. A, 2001, vol. 32A, pp. 2905–08.
X.V. Eynde, J.P. Servais, M. Lamberigts: Surf. Interface Anal., 2003, vol. 35, pp. 1004–14.
J. Mahieu, S. Claessens, B.C. De Cooman, and F. Goodwin: The 6th International Conference on Zinc and Zinc Alloy Coated Steel Sheet (GALVATECH 2004), Chicago, Illinois, 2004, Association for Iron and Steel Technology, pp. 529–38.
Y.F. Gong, H.S. Kim, B.C. De Cooman: ISIJ Int., 2008, vol. 48, pp. 1745–51.
Y.F. Gong, H.S. Kim, B.C. De Cooman: ISIJ Int., 2009, vol. 49, pp. 557–63.
L. Cho, M.S. Kim, Y.H. Kim, S.J. Lee, and B.C. De Cooman: Proceedings of the 8th International Conference on Zinc and Zinc Alloy Coated Steel Sheet (GALVATECH 2011), Genova, Italy, 2011, Associazione Italiana di Metallurgia, pp. 145–52.
X.S. Li, S.I. Baek, C.S. Oh, S.J. Kim, Y.W. Kim: Scripta Mater., 2007, vol. 57, pp. 113–16.
A.R. Marder: Prog. Mater. Sci., 2000, vol. 45, pp. 191–271.
B. Mintz: Int. Mater. Rev., 2001, vol. 46, pp. 169–97.
L. Chen, H.S. Kim, S.K. Kim, B.C. De Cooman: ISIJ Int., 2007, vol. 47, pp. 1804–12.
C.E. Jordan, K.M. Goggins, A.R. Marder: Metall. Mater. Trans. A, 1994, vol. 25A, pp. 2101–09.
I. Hertveldt, B.C. De Cooman, S. Claessens: Metall. Mater. Trans. A, 2000, vol. 31A, pp. 1225–32.
S. Feliu, M. Pérez-Revenga: Acta Mater., 2005, vol. 53, pp. 2857–66.
M. Blumenau, M. Norden, F. Friedel, K. Peters: Steel Res. Int., 2010, vol. 81, pp. 1125–36.
C. Wagner: Z. für Elektrochem. Ber. Bunsenges. Phys. Chem., 1959, vol. 63, pp. 772–82.
G. Böhm, M. Kahlweit: Acta Metall., 1964, vol. 12, pp. 641–48.
R.A. Rapp: Corrosion, 1965, vol. 21, pp. 382–401.
Y. Niu, F. Gesmundo: Oxid. Met., 2006, vol. 65, pp. 329–55.
D. Huin, P. Flauder, J.B. Leblond: Oxid. Met., 2005, vol. 64, pp. 131–67.
J.B. Brunac, D. Huin, J.B. Leblond: Oxid. Met., 2010, vol. 73, pp. 565–89.
R. Rapp: Acta Metall., 1961, vol. 9, pp. 730–41.
L. Cho, S. Lee, M. Kim, Y. Kim, B.C. De Cooman: Metall. Mater. Trans. A, 2013, vol. 44A, pp. 362–71.
J. Takada, M. Adachi: J. Mater. Sci., 1986, vol. 21, pp. 2133–37.
H. Oikawa: Technology Reports, Tohoku University, 1983, vol. 48, pp. 7–77.
D.R. Lide: CRC Handbook of Chemistry and Physics, 86th ed., CRC Press, Boca Raton, 2005.
L. Cho, M.S. Kim, Y.H. Kim, B.C. De Cooman: Metall. Mater. Trans. A, 2013, vol. 44A, pp. 5081–95.
H. Liu, Y. He, L. Li: Appl. Surf. Sci., 2009, vol. 256, pp. 1399–1403.
H. Liu, Y. He, S. Swaminathan, M. Rohwerder, L. Li: Surf. Coat. Technol., 2011, vol. 206, pp. 1237–43.
Y. Suzuki, T. Yamashita, Y. Sugimoto, S. Fujita, S. Yamaguchi: ISIJ Int., 2009, vol. 49, pp. 564–73.
N. Birks, G.H. Meier, and F.S. Pettit: Introduction to the High Temperature Oxidation of Metals, 2nd ed., Cambridge University Press, New York, 2006, pp. 114–15.
J.M. Mataigne, M. Lamberigts, and V. Leroy: Developments in the Annealing of Sheet Steels, The Minerals, Metals and Materials Society (TMS), Warrendale, PA, 1992, pp. 511–28.
F. Wang: Oxid. Met., 1997, vol. 48, pp. 215–24.
Z. Liu, W. Gao, K.L. Dahm, F. Wang: Acta Mater., 1998, vol. 46, pp. 1691–700.
S. Guan, W. Smeltzer: Oxid. Met., 1994, vol. 42, pp. 375–91.
J. Töpfer, R. Dieckmann: Solid State Ionics, 2010, vol. 181, pp. 479–88.
I. Burn, S. Neirman: J. Mater. Sci., 1982, vol. 17, pp. 3510–24.
Acknowledgments
The authors gratefully acknowledge the support of Dr. Myung Soo Kim and Dr. Young Ha Kim of the POSCO technical Research Laboratories, Gwangyang, South Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted October 3, 2013.
Appendix: Application of Huin’s Model for Numerical Simulation
Appendix: Application of Huin’s Model for Numerical Simulation
Huin’s model[19] was used to calculate the internal oxidation of a ternary Mn-Si-Fe alloy where Fe is regarded as a matrix. During an isothermal annealing, Fe remains inert, whereas Mn and Si form MnO and SiO2, respectively. Numerical simulation was carried out by solving the following diffusion equations for Mn, Si, and oxygen.
Here, F Mn, F Si, and F O are the total mass fractions of Mn, Si, and oxygen (in all its possible forms, that is dissolved in the matrix or bound to precipitates), in ppm, respectively. C Mn, C Si, and C O are the dissolved mass fractions of Mn, Si, and oxygen, in ppm, respectively.
Several conditions should be considered for the calculation. First, counting atoms of element in all their possible forms (dissolved in the matrix or bound to precipitates) yields the following mass balance equations.
P MnO and \( P_{{{\text{SiO}}_{ 2} }} \) are the mass fractions of the precipitates of MnO and SiO2, in ppm, respectively. M Mn, M Si, and M O are the atomic mass of Mn, Si and oxygen, respectively. M MnO and \( M_{{{\text{SiO}}_{ 2} }} \) are the molecular mass of MnO and SiO2, respectively.
The expression for the local thermodynamic equilibrium governed by the laws of mass action yields the following equations:
Here, K MnO is the solubility product of MnO, in (ppm)2. \( K_{{{\text{SiO}}_{ 2} }} \) is the solubility product of SiO2, in (ppm)3. Equation [8] indicates that there will be no precipitation when the product of C Mn and C O is less than the value of K MnO. When the product of C Mn and C O exceeds the value of K MnO, C Mn and C O will react until the product of C Mn and C O equals to the value of K MnO. A similar relation holds in the case of SiO2.
The one-dimension problem is considered, with the spatial coordinate, the depth below the free surface, being denoted x. All quantities at time t (denoted with an upper index(0)) being assumed to be known, the problem is to determine all quantities at time t + ∆t (denoted without any index). The spatial discretization involving N nodes x 1 = 0, x 2, …, x N is introduced (Figure 14). In practice, node 1 will represent the outer surface of the sheet in contact with the annealing atmosphere, and node N will be located far below the surface.

Spatial discretization[19]
Discretization of the ordinary differential equations (Eq. [6]) was done by finite difference method using an explicit Euler scheme order 1, as given in Eq. [10].
Here, the time-step, ∆t, is taken as 1/10 of the quantities (∆x k )2/D O for the sake of numerical accuracy. These equations do not apply at the boundary nodes x 1 and x N . At node x 1, Eq. [11] is adopted instead of (10) and the theoretical basis on the treatment of the boundary condition is described in the Huin’s original work[19,20]:
At node x 1, if it is assumed that chemical equilibrium is rapidly achieved, the dissolved oxygen at the surface, \( C_{\text{O}}^{{ ( {\text{S)}}}} \), is obtained by combining Eqs. [2], [3], and [4]:
At node x N , deep in the steel sheet, it is assumed that the Mn and Si concentrations remain constant at their nominal value given by the steel composition, and that no MnO and SiO2 are present:
Here, \( C_{\text{Mn}}^{{ ( {\text{O)}}}} \) and \( C_{\text{Si}}^{{ ( {\text{O)}}}} \) are the Mn and Si mass concentrations in the steel.
Finally, if all the values of F Mn(x k ), F Si(x k ), F O(x k ), C Mn(x k ), C Si(x k ), C O(x k ), P MnO(x k ), and \( P_{{{\text{SiO}}_{ 2} }} \)(x k ) at every node are known at the time t, the calculation of the values at t + ∆t can be carried out by following sequences:
-
1.
At node x 1, F Mn(x 1) and F Si(x 1) are obtained by Eq. [11]. C Mn(x 1) and C Si(x 1) then follow by solving the mass balance equations (Eq. [7]). P MnO(x 1 ) and \( P_{{{\text{SiO}}_{ 2} }} \)(x 1) are calculated by thermodynamic equilibrium (Eqs. [8] and [9]) as the term C O(x 1) in Eqs. [8] and [9] is already known by Eq. [12]. F O(x 1) is finally obtained by Eq. [7].
-
2.
At nodes x 2, x 3, …, x N , the precipitation of SiO2, which has a low solubility product, is considered prior to that of MnO. F Si(x k ) is obtained from the finite difference method (Eq. [10]). If SiO2 precipitate, none-zero \( P_{{{\text{SiO}}_{ 2} }} \) can be obtained by the calculation of thermodynamic equilibrium. Because the C Si(x k ) and C O(x k ) terms in Eq. [9] are unknown, P MnO should be obtained by expressing C Mn(x k ) and C O(x k ) in terms of F Mn(x k ) and F O(x k ) using Eq. [7]. This yields Eq. [14].
$$ \left( {F_{\text{Si}} - \frac{{M_{\text{Si}} }}{{M_{{{\text{SiO}}_{ 2} }} }}P_{{{\text{SiO}}_{ 2} }} } \right) \times \left( {F_{\text{O}} - \frac{{M_{\text{O}} }}{{M_{\text{MnO}} }}P_{\text{MnO}} - 2\times \frac{{M_{\text{O}} }}{{M_{{{\text{SiO}}_{ 2} }} }}P_{{{\text{SiO}}_{ 2} }} } \right)^{ 2} { = }K_{{{\text{SiO}}_{ 2} }} . { } $$(14)Equation [14] can be solved numerically using Newton–Raphson method. C Si(x k ) and C O(x k ). This is followed by solving Eq. [7]. In the same manner, F Mn(x k ) is obtained from Eq. [10]. Non-zero values of P MnO can be obtained by Eqs. [7] and [8], which yields Eq. [15].
$$ \left( {F_{\text{Mn}} - \frac{{M_{\text{Mn}} }}{{M_{\text{MnO}} }}P_{\text{MnO}} } \right) \times \left( {F_{\text{O}} - \frac{{M_{\text{O}} }}{{M_{\text{MnO}} }}P_{\text{MnO}} - 2\times \frac{{M_{\text{O}} }}{{M_{{{\text{SiO}}_{ 2} }} }}P_{{{\text{SiO}}_{ 2} }} } \right) = K_{\text{MnO}} . { } $$(15)Equation [15] is solved numerically using Newton–Raphson method. C Mn(x k ) and C O(x k ) are then updated by the mass balance equations.
-
3.
At node x N , all the parameter values are kept constant, as expressed in Eq. [13].
Rights and permissions
About this article

Cite this article
Cho, L., Jung, G.S. & De Cooman, B.C. On the Transition of Internal to External Selective Oxidation on CMnSi TRIP Steel. Metall Mater Trans A 45, 5158–5172 (2014). https://doi.org/10.1007/s11661-014-2442-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-014-2442-7