
Abstract
Grain boundary sliding (GBS) is an important deformation mechanism for elevated temperature forming processes. Molecular dynamics simulations are used to investigate the effect of solute atoms in near grain boundaries (GBs) on the sliding of Al bicrystals at 750 K (477 °C). The threshold stress for GBS is computed for a variety of GBs with different structures and energies. Without solute atoms, low-energy GBs tend to exhibit significantly less sliding than high-energy GBs. Simulation results show that elements which tend to phase segregate from Al, such as Si, can enhance GBS in high-energy GBs by weakening Al bonds and by increasing atomic mobility. In comparison, intermetallic forming elements, such as Mg, will form immobile Mg-Al clusters, decrease diffusivity, and inhibit GBS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Grain boundary sliding (GBS) involves the rigid translation of one grain over another adjacent grain parallel to the grain boundary (GB) interface. GBS mediates the plastic flow of polycrystalline materials,[1,2] especially when grain sizes decrease to nanometer scale.[3] For micron-sized grains, GBS becomes an important deformation mechanism for elevated temperature forming, such as quick plastic forming or superplastic forming.[4–12] It is important to understand how to modify the chemistry at GBs through solute alloy additions to increase the contribution of GBS to total deformation so that the extra processes for refining grain sizes can be avoided. Extensive experimental evidence has suggested that GB chemistry plays an important role in controlling GBS. For example, Vetrano et al.[13–15] have suggested that Sn segregates to GBs and enhances GBS in polycrystalline Al-Mg alloys. In these materials, the Sn in the GB is considered to be a separate phase with lower melting temperature, which decreases the shear strength of the GB. McFadden and Mukherjee[16] showed that superplasticity cannot be achieved in Ni without sulfur, and concluded that the role of sulfur in Ni superplasticity is to inhibit grain growth and enhance GBS by weakening Ni-Ni bonds. The effect of the solute atoms on GBS likely depends on their concentration in the alloy, their segregation level at the GB, and their ability to enhance or inhibit GBS.
Many alloying elements in superplastic alloys indeed segregate to GBs, either during static aging or deformation. In commercial-purity Al AA5083 alloys, Si segregates to the GBs, and the concentration of Si at the GBs increases with strain and decreases with deformation temperature.[13] Li et al.[17] showed that ultrasonic melt treatment can produce a uniform distribution of Si segregation to the Al GBs. The segregation of Sn and Zr to GBs also has been observed in a polycrystalline Al-Mg-Mn-Sn alloy during static ageing at 723 K (450 °C) to 823 K (550 °C).[13,14] Mg segregated to the GBs during static aging[18] but depleted during plastic deformation.[13,14] Computational studies have confirmed these observations. For example, Liu et al.[19,20] have performed molecular dynamic simulations of Al-Mg alloys using the embedded atom method (EAM) potential. Their results show that Mg will segregate to GBs and that the level of segregation depends on the structure of the GB; the Mg segregation level is higher near lower angle [110] tilt boundaries but shows minimal variations at [100] twist boundaries. They also found that Mg segregation will reduce the decohesion energy at Al GBs. Similar observations of the Mg solute effect were made by Liu et al.[21] using more accurate first-principle calculations at a Σ11(113) GB.
The understanding of how an individual alloying element affects GBS is rather limited. Most experimental data are measured indirectly from polycrystals, bicrystals, or tricrystals of alloys, and the conclusions are sometimes contradictory. For example, various conclusions have been made for the commonly used Cu additions in Al alloys. Green et al.[22] measured an increase in creep rate from the acceleration of GBS by Cu additions to an AA5083-based polycrystalline Al alloy, suggesting that Cu enhances GBS. Weinberg[23] and Halliday and Beevers[24] found that a small amount of Cu (0.05 pct ~ 0.3 pct) had no effect on GBS. Both Kato[25] and Mima et al.[26] studied sliding in bicrystals of Al-Cu solid solutions and noted that small Cu additions reduced GBS. The influence on GBS by other alloying elements in Al alloys, including Mg, Mn, Zn, Si, and Fe, are available from scattered experimental data. Mg (0.05 pct to 1.8 pct) first reduced sliding at small concentrations then enhanced sliding at higher concentrations.[26] Mima et al.[26] compared bicrystals of Al individually alloyed with Mn (0.02 pct to 0.76 pct) or Zn (0.07 pct to 8.1 pct) and observed that Mn reduced GBS more than Zn at small concentrations. Weinberg[23] found that Si (0.10 pct) had no effect on GBS with tricrystals experiments. In this same study, Weinberg[23] found that 0.08 pct Fe in aluminum inhibited GBS. These experimental results do not provide guidelines for engineering GBs to enhance GBS. Atomic simulations also have been used to understand how individual solute elements affect GBS. Namilae et al.[27] studied the sliding of symmetric tilt Al GBs with EAM potential of Mg-Al and showed that the presence of Mg at the GB increased GBS slightly in some GBs. The current lack of understanding of the effect of solute elements on GB severely limits our ability to engineer new alloys with enhanced GBS and improved formability.
In this article, we use molecular dynamic (MD) simulations to study the effect of solute atoms that already have segregated to GBs, on the sliding behavior of Al GBs at 750 K (477 °C). More specifically, we focus on the effect of solute atoms on the threshold stress for GBS. The existence of a threshold stress for GBS has been postulated by Ashby and Verrall[28] and Gittus,[29] and proved in experiments[30] and atomistic simulations.[31] Microstructure-based simulations show that the flow stress of polycrystalline Al alloys deformed at a low strain rate (below 10−3 seconds−1) is sensitive to small changes in the threshold stress.[32] Thus, understanding the role of GB structure on the threshold stress is of particular interest. The threshold stress decreases with increasing temperature[33] and with increasing GB energy.[31] Because the low energy and the special low-Σ coincidence boundaries are common in these commercial superplastic materials,[34] we will compare the vacancy effect on both low energy, symmetric tilt Σ3 GB and several other high energy asymmetric tilt GBs in this study.
In this study, we compare the effects of two common alloying elements, Mg and Si, in Al. Their different phase diagrams with Al reflect their drastically different atomic interactions when alloying with Al. Si phase segregates from Al, forming eutectic alloys, and Mg forms several intermetallic phases. The solubility of Si in Al is about 1.6 at pct, much smaller than 16.3 pct for Mg. In terms of atomic radius, Si is smaller than Al, and Mg is larger than Al.[35] So, understanding their difference in changing GBS threshold stress at Al GBs may suggest how to select other alloy elements from basic and measurable material properties, such as the phase diagram with Al, the atomic size, the heat of mixing, and the heat of solution in Al. The phase segregation of Al-Si indicates that the heat of formation of Al-Si is positive, and the existence of several Al-Mg intermetallics suggests that their heats of formation are negative. Our simulations use the modified embedded atom method (MEAM) potentials to mimic the relative heats of formation of Al-Si and Al-Mg alloys correctly. We find that Si impurities greatly decrease the sliding threshold in high-energy GBs by weakening Al bonds but have less influence on low-energy GBs. In contrast, Mg impurities increase sliding threshold of all GBs by forming immobile Mg-Al clusters. This clustering occurs because of the negative heat of formation of Mg with Al.
2 Method of Simulations
2.1 Atomic Potential
The interactions between Al atoms are described by the MEAM potential and are reparameterized to include the second nearest neighbors.[36] The new parameters in the MEAM potential correctly predict face centered cubic (fcc) as the lowest energy phase for Al, predict Al surface energies are in good agreement with first-principles calculations, and predict the melting temperature correctly,[31] which is important for high-temperature simulations. The MEAM potential describing the interactions of Al-Si and Al-Mg is developed in References 37 and 38, respectively. MEAM potential is similar to the more common EAM potentials but allows for the directional dependence of Si bonding.
MEAM defines the energy E of the system as the sum of energies for each atom i, with each atom having energy contributions from an embedding function F that depends on a local electron density \( \bar{\rho }_{i} \) and a pair potential ϕ as follows:
The pair potential only depends on the distance between atoms i and j, \( R_{ij} = \left| {\vec{r}_{i} - \vec{r}_{j} } \right|, \) and the form of the embedding function F is expressed as follows:
where A is a constant, E c is the cohesive energy, and \( \bar{\rho }^{o} \) is a reference electron density. The actual angular dependence of the potential is buried in the definition of \( \bar{\rho }, \) which represents the electron density contributions from a spherically symmetric term and three angularly dependent terms.
To describe an alloy system, the pair potential ϕ between different elements was given as the energy difference with respect to the total energy of a hypothetical ordered structure (reference structure). Because most parameters and fitting procedures are given in References 37 and 38, we only list the key parameters used for Al-X (X = Si or Mg) interactions in Table I. In the potentials used here, both Al-Mg and Al-Si interactions were constructed referring to the rock-salt or B1 structure. The cohesion energy for the reference B1 structure of Al-X (where X either can be Mg or Si) is defined as \( E_{C}^{Al - X} = \left( {E_{c}^{Al} + E_{c}^{X} } \right)/2 - \Updelta E, \) where ΔE is the heat of formation for the reference B1 structure. The heat of formation of other phases can be calculated from ΔE. We believe that the main difference between Al-Si and Al-Mg interactions is that Si has a positive heat of formation with Al, whereas Mg has negative heat of formation in several phases. Reference 37 listed ΔE = 0.255eV/atom for Al-Si, corresponding to the experimentally observed Al-Si phase segregation. In Reference 38, ΔE for Al-Mg is listed as +0.4575 eV/atom. This indicates a large positive heat of formation of +0.4575 eV/atom for the Al-Mg B1 phase, and consequently leads to a positive heat of formations for all Al-Mg intermetallic compounds, including a positive heat of formation (~0.4 eV) for the Mg17Al12 phase. This finding is inconsistent with the experimental observation that Mg17Al12 phase has a small negative heat of formation (−0.017 eV from density functional theory [DFT] calculations). Furthermore, the parameters used in Reference 38 would predict that Mg segregates more than Si from Al, which does not capture the major difference between Mg and Si when alloying with Al. So we simply use ΔE =−0.4575 eV/atom for Al-Mg in our calculations. This change will exaggerate the negative heat of formation for Mg. Because this change still cannot match perfectly all properties for Al-Mg alloys, we refer it as pseudo Mg atom Mg* instead of real Mg atoms. Because the parameters used for Si are suitable for Al-Si systems, we still can refer to it as Si. However we believe that, with the set of parameters listed in Table I, our simulations can be used to compare the behavior of alloying elements that either phase segregate from Al (Si) or form ordered phases with Al (Mg*).
2.2 GBS Simulation Setup
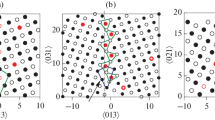
Six different bicrystal structures are investigated in this work, including one symmetric Σ3(111)[110] tilt GB and five asymmetric incommensurate [110] tilt GBs with misorientation angles of 10 deg, 25 deg, 35 deg, 55 deg, and 90 deg. In the coincident-site-lattice notation, the Σ3(111)[110] tilt GB is formed by rotating one of the two Al grains about the [110] tilt axis by 60 deg. The boundary is oriented along the (111) plane. The Σ3(111)[110] GB of Al is simulated by the orthorhombic supercell shown in Figure 1(a). The supercell contains 2100 atoms distributed in 50 (111) layers parallel to the GB. The dimensions of the supercell along the \( \left\langle {111} \right\rangle ,\left\langle {11\bar{2}} \right\rangle , \) and \( \left\langle {1\bar{1}0} \right\rangle \) directions are 100.00 Å, 34.78 Å, and 17.21 Å, respectively. The two grains in the asymmetric bicrystal structures have parallel \( \left\langle {1\bar{1}0} \right\rangle \) directions that lie in the plane of the GB (the yz plane). One grain is rotated around this \( \left\langle {1\bar{1}0} \right\rangle \) axis (the z axis) to generate tilt GBs with mismatch angles of 10 deg, 25 deg, 35 deg, 55 deg, and 90 deg, (as shown in Figure 1(b)). Each GB structure is nonperiodic in the x-direction, which is perpendicular to the GB plane and is periodic in the GB plane along the y- and z-directions. Because the crystalline orientation along the y direction is different for two grains, to maintain the periodic boundary condition, a common periodic cell length was chosen (more detailed cell geometry can be found in Reference 31). Each grain is about 56 Å thick along the x direction.

The following GB structures prior to relaxation: (a) symmetric tilt Σ3 GB (b) asymmetric tilt GB with rotation angle θ, where θ can be 10 deg, 25 deg, 35 deg, 55 deg, and 90 deg. Each GB structure is divided into the following zones along the x-direction: fixed zone (the outmost layers on both sides), the force-applying zone (indicated by the dash-dot boxes), and the free zone (all other atoms)
Based on the as-built GBs, some atoms in the GB region are selected randomly and replaced by substitution impurities. The GB region is defined as a layer about 10 Å thick containing the GB plane. This region usually contains about two to three layers of atoms on each side of the GB plane, depending on the specific GB structure. For each GB structure, solute atoms (Mg* or Si) with concentrations of 2 pct and 15 pct, respectively, randomly are introduced into the GB region. The concentration is defined by the number of solute atoms normalized by the number of atoms at the GB zone with 10 Å width. After the GB structures with solute atoms were created, they were relaxed using a constant temperature, constant stress ensemble until they reached the equilibrium temperature and zero stress. In this process, the structures first were relaxed at 300 K (27 °C) for 25 ps, with 1 fs time step. During the process, both the local positions of the atoms and the periodic length of the simulation box in y- and z-directions are relaxed. The equilibrium structures at 300 K (27 °C) then are relaxed again at 750 K (477 °C), which is the desired temperature for GBS for 25 ps. The total energy of the simulation cell usually converges within 5 ps, and the GB structures are in a local equilibrium state by the end of the two-step relaxation.
GBS along the \( \left\langle {1\bar{1}0} \right\rangle \) direction is studied for all GB structures. We picked the sliding direction along the tilt axis because this direction is easy sliding, and it will have a higher probability of sliding under complicated loading in a polycrystal; thus, it is more representative. The simulations of GBS are conducted at 750 K (477 °C) with nonequilibrium molecular dynamics. The temperature is kept constant at 750 K (477 °C) with a Nose–Hoover thermostat.[39–41] The simulation cell is kept constant. The two grains are sheared by applying forces to the atoms near the top and bottom surfaces of the cell. As shown in Figure 1, constant forces in the z-direction \( \left( {\left\langle {1\bar{1}0} \right\rangle } \right) \) are applied to the atoms inside the two boxes (the moving zone). In addition, the displacement of the outermost one to two atomic layers along the nonperiodic (x) direction is constrained to prevent free surface melting at high temperatures and large vibrations of the structure in the x-y plane. These boundary conditions induce a shear stress on the GB. The applied shear stress is computed by multiplying the applied atomic forces by the number of atoms in the moving zone, divided by the total number of atoms and the GB area. The time step is 1 fs. Simulations with progressively increasing applied shear stress were carried out for each GB, and the relative displacement of the two grains was recorded as a function of time. The threshold stress for GBS was determined using small brackets of applied shear stress. A GB is assumed to slide if the relative displacement of the two grains exceeds 5 Å. On rare occasions, some low-energy GBs initially showed sliding but came to rest when the GB reached a second equilibrium configuration. This pattern was classified as no sliding.
3 Results and Discussion
First, the overall effect of Si and Mg* solute atoms on the threshold stress for GBS of Al GBs is shown in Figure 2 and tabulated in Table II. Previous MD simulations[31] of pure Al GBs have revealed that the constant GBS velocity increase linearly with the applied stress as \( v_{t} = \left( {\sigma_{t} - \sigma_{c} } \right)/\eta , \) where σ c is the threshold stress, below which no sliding occurs. The value of the threshold stress for GBS decreases with increasing GB energy. GBs with lower energy have a higher threshold stress and, thus, an increased resistance to sliding. The GB energy was calculated at 300 K (27 °C), and the GBS threshold stress for the six pure GBs (referring to GB without solute atoms) are listed in Table II for comparison. The threshold stress is plotted as a function of the corresponding pure GB energies calculated at 300 K (27 °C) in Figure 2. The results show that Si solutes significantly decrease the threshold stress for GBS in the three lower energy GBs and have less effect in the higher energy GBs. The effect of Si impurities is enhanced with an increased concentration of Si. For example, the threshold stress of the symmetric tilt Σ3 boundary is reduced from 1.2 GPa by 0.1 GPa with 2 pct Si solute atoms and by 0.9 GPa (more than half of the original value) with 15 pct Si solute atoms. The decrease of GBS threshold stress with Si solute atoms is not significant in the high-energy boundaries, such as those with 35 deg, 55 deg, and 25 deg misorientations. The effect of Si on GBS is negligible for high-energy GBs because of the already high atom mobility in these boundaries. In comparison, Mg* solutes increase the threshold stress in all six GBs by more than 0.1 GPa for all boundaries. The increment of threshold stress does not increase with higher Mg* concentrations.

Effect of solute atoms of (a) Si and (b) Mg* at GB region on GBS threshold stress in all six GBs
3.1 GB Mobility and Sliding
To understand how the segregated solute atoms at GBs change GBS, we investigate the sliding behavior and the atomic mobility in more detail for the special Σ3 GBs with or without 15 pct Si and Mg* solute atoms in the GB region. Figure 3 plots the relative tangential displacement of the two grains as a function of time for pure and solute atom doped Σ3 GBs subjected to the same resolved shear stress (0.4 GPa). The fluctuating relative tangential displacement between the two grains indicates that this shear stress is not sufficiently large to result in GBS in either the pure GB structure or the boundary doped with Mg*. In contrast, the same applied shear stress leads to GBS when the GB is doped with 15 pct Si atoms.

Relative tangent displacement of two grains as a function of time for Σ3 GBs
Recently, high-resolution transmission electron microscopy has observed a less than 1-nanometer-thick premelted GB structure, especially in metals with strongly segregated impurities, such as Ni-doped W[42,43] and Ni-doped Mo.[44] These systems are similar to Si-doped Al GBs. Because of the Si addition atomic disordering at the boundary can be observed during GBS, but this does not provide direct evidence of an increase in the atomic mobility at the GB. To capture the effect of solute atoms on the GB mobility and atomic diffusion, we computed the atomic root-mean-squared-displacement (RMSD) for each atom i, as follows:
where \( \vec{r}_{i} \left( t \right) \) are the position vector of atom i at time t, Δt is the time interval, T is the total simulation time, and < > denotes an average across the time frames during the total simulation time of T = 50 ps. The x and y components of the RMSD do not contain the sliding motion and provide a measure of average atomic displacement and hopping distance during time interval Δt, which was set to be 1 ps. For most atoms in the bulk, the atomic displacement is purely vibration during 1 ps. However, if atoms are melted or diffuse away from their equilibrium positions during this time interval, then they will exhibit much larger RMSD. The Lindemann criterion[45] relates the origins of melting to the atomic mobility indicated by RMSD. When the thermal RMSD of the atoms in the lattice reaches a critical fraction of the equilibrium interatomic distance, (typically ~10 pct to 15 pct for the bulk and ~25 pct for the surface[46]), those atoms are considered to be melted. Previous GBS simulations have proved that when RMSD exceeds 0.7 Å in Al, it indicates premelting at GBs.[31]
To compute the RMSD on a representative y-z plane (parallel to GB), the atoms were partitioned into smaller slabs (2-Å-thick) parallel to the GB. Figure 4 shows the variation of RMSD on the y-z plane as a function of the distance of the center of the slab from the base of the simulation cell. The GB position is about x = 50 Å. The atoms that are away from the GB plane and at the center of the grain exhibit the same RMSD value of ~0.3 Å for all GBs, indicating the atoms inside each grain only vibrate around their equilibrium positions. Figure 4 compares RMSD distributions for the same GBs with different solute atoms and under the same applied shear stress (0.4 GPa). The GB with 15 pct Si solute atoms has much higher RMSD in the GB vicinity than pure Al Σ3 GB, indicating the higher atomic mobility at GB. In contrast, the 15 pct Mg solute atoms doped GB has the lowest RMSD and, therefore, the lowest atomic mobility and diffusivity at GB. The increase of atomic mobility correlates with a decrease in the GBS threshold stress, and the decrease of atomic mobility will increase the GBS threshold stress, as shown in Figures 2(a) and (b). Thus, the solute atoms seem to be able to influence GBS by changing the atomic diffusivity at the GB. In previous studies, we have demonstrated that the RMSD at the GB plane is much lower at Σ3 GB than those in high-energy asymmetric tilt GBs. The atomic RMSD also increases with the applied shear stress, indicating that GB premelting occurred beyond a critical shear stress.[31] Doping GB with vacancies can increase the mobility of the atoms at the GB and induce GB premelting, thus lowering GBS threshold stress.[47] Si impurities seem to enhance GBS by a similar process.

The atomic RMSD at Σ3 GBs during GBS as a function of the distance to the GB plane, which is noted by the vertical dash line
3.2 Atomic Bonding with Solute Atoms
The changes of atomic diffusivity and GBS produced by solute atoms are a direct result of the changes of the nature of solute-Al atomic bonds. To further illustrate how the solute atoms interact with Al atoms, the bond distances between the solute atom and its nearest neighbors (12 Al atoms) before sliding are tracked during GBS. The bond distance between the solute atom and one of its neighbors d is normalized by the Al-Al bond distance (or the nearest neighbor distance) in pure Al d 0 . We then average all 12 bonds for all solute atoms and follow the change in d/d 0 ratio during the simulation time while the GBs are sliding. As a representative result, Figure 5 shows the d/d 0 ratio for GBs with a 10 deg misorientation angle and with 15 pct Si or Mg* solute atoms subjected to the same shear stress of 0.71 GPa. Under this shear stress, both GBs slide. At the beginning, the d/d 0 ratio is close to 1. Because Si is smaller than Al and because Mg is larger than Al, we can observe that the d/d 0 ratio is slightly below 1 for Al-Si and is slightly above 1 for Al-Mg*. As GBS takes place in both Si- and Mg*-doped GBs, the d/d 0 ratio increases for Si-Al bonds, whereas the d/d 0 ratio remains close to 1 for Mg-Al bonds during GBS. After 10 ps, the average distance between Si solute atoms and their initial bonded neighbors is about twice the original Al-Si bond distance. The increasing distance between Si solute atoms and their original bonded Al neighbors indicates that the Al-Si bonds break during GBS. In contrast, the constant value of Mg* solute and their bonded Al neighbors indicates that no Mg*-Al bonds are broken during GBS. Individual Mg* solute atoms cluster with at least their 12 nearest neighbors, forming obstacles for GBS and, thus, increasing the threshold stress for GBS. In Al-Mg alloys, intermetallics could form around GBs, which might resist GBS in a similar fashion as the small Al-Mg clusters.

Average distance between impurities and their neighboring Al atoms during sliding at the GB with a misorientation angle of 10 deg with 15 pct impurities and under the shear stress = 0.71 GPa
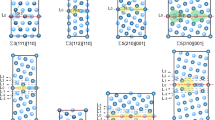
Snapshots showing the detailed atomic structures of the GBs containing Si and Mg* solute atoms, before and after 25 ps of sliding, are plotted in Figure 6. After atomic relaxation, before GBS, we highlighted the GB based on the atomic orientations of the two grains. Note that the GB structures are not a flat plane after relaxation; impurity atoms create GB steps and ledges. Although the two grains slide in a rather continuous fashion, not all the atoms move together. Tracking local atomic units with about 12 atoms, we found these local atomic units stick or slip at different times. Thus, GBS at a high temperature is mitigated by local atomic rearrangement or atom shuffling. Alloying additions can change the local atomic shuffling by changing local atomic interactions, as shown in Figure 5. Although the details of these local events are interesting, the alloying effect on GBS threshold stress is consistent with different randomness of the impurity locations. Figure 6 also shows the structures after GBS, which reveal that sliding occurs at different places. GBS is localized in the atomic layers containing Si atoms at the Si-doped GBs; in comparison, GBS occurs between the atomic layers that contain no Mg* at the Mg*-doped GBs. These results clearly show that Mg* impurities form immobile particles with the surrounding Al atoms during GBS. The addition of Mg* also induced slight GB migration. Although GB shear and migration coupling is not dominant at a high temperature,[33] this observation suggested that impurity atoms can influence GBS and migration coupling, which will be subject to future research.

(color online) Snapshot of asymmetric tilt GB with misorientation angle of 10 deg. (a) GB doped with 15 pct Si under the shear stress = 0.71 GPa before and after GBS; (b) GB doped with 15 pct Mg under the shear stress of 0.71 GPa before and after GBS. The green line highlighted the location of the GB based on atomic orientations. The orange color is for Si atoms, and the red color is for Mg atoms
This atomic bond change in molecular dynamics simulations occurs because of the empirical atomic interactions we employed; Si has a much weaker bond with Al, but Mg* forms a stronger bond with Al. In atomic simulations, the energy of individual atoms signifies the stability of the atoms and its neighbors. Figure 7 shows the atomic energy of Al, defined in Eq. [1], for GBs with a 10 deg misorientation angle. Contour plots are shown of atomic energy for pure GBs and GBs with 15 pct Si or Mg* for the fully relaxed GB structure. The solute atoms are removed from the plots to illustrate the change in energy of Al atoms near the GB. The plots show that the Al atoms near the Si solute atoms have higher energy than those in pure Al GBs. They also show that the energies of atoms in the GBs with Mg* solutes are lower compared with those in the pure Al GB. This energy change is mainly a result of the change from the pair potential term of atomic energy defined in MEAM. More specifically, the pair potential change is mainly from ΔE, when an Al atom interacts with Si or Mg*. As is shown, the average atomic energies on Al atoms with Si addition increase about ~0.13 eV from −3.15 eV to −3.02 eV, and decrease about ~0.20 eV from −3.15 eV to −3.34 eV with Mg* addition. The atomic energy change on Al is about half of the ΔE value, as it is 0.255 eV for Si-Al interaction and −0.45 eV for Mg*-Al interaction. One consequence of the atomic energy change can be understood as when the atomic energy increases with solute addition, the atomic interaction is weakened, and the atomic mobility and diffusivity at GBs will be increased, thus enhancing GBS by reducing GBS threshold stress and vice versa.

(color online) Contour plot of atomic energy of the grain boundary with misorientation angle of 10 deg with (a) pure GB, (b) 15 pct Si, and (c) with 15 pct Mg. Note that the solute atoms are deleted from the structures. The color scales indicate the atomic energy
3.3 Discussion
Although the Si and Mg* atoms have different sizes compared with Al atoms, the change of atomic interaction because of solute atoms suggests that the chemistry effect is more important than size in influencing GBS. The size difference alone cannot account for the dramatic differences in GBS associated with the two solutes. Our simulation results demonstrate that the threshold stress for GBS is correlated to the increase in energy of Al atoms near the GB. The energy change of Al atoms around the solute atoms suggests that the Si solute atoms increase the mobility of surrounding Al atoms by increasing their energy level and that Mg* decreases the mobility of the surrounding Al atoms by reducing their energies. This occurs because the Al-Mg* bond is much stronger than the Al-Si bond. This conclusion is consistent with the effect of S in Ni on the superplasticity of Ni. DFT calculations show that the presence of S weakens Ni-Ni GBs.[48] The reduced bond strength across the GBs is believed to enhance GBS at an elevated temperature leading to experimentally observed superplasticity in Ni-S alloys.[16] Our simulations suggest that adding strong phase segregation alloys, such as Si in Al, can increase the mobility of the atoms at the GB and induce GB premelting, which agree well with recent experimental evidence on Ni-doped W[42,43] and Ni-doped Mo[44]—both are strong segregation systems. The weak segregation alloys elements, such as Mg in Al, tend to have higher solubility in the bulk and less concentration at the GB region. In our simulations, we set the same solute concentrations to facilitate the comparison of their chemical influences on GBS. It seems, if weak segregation alloys reach a high enough concentration at the GB region, then the tendency of GB premelting will be lowered and the atomic motions at the GB will slow down. This finding is consistent with recent experiments and simulations of a weak segregation Ni-W system in which W is believed to stabilize nanocrystalline structures[49–54] by stabilizing GB. The GB structures for alloys are more complex than that of pure metals and, thus, require new theories that treat GB as a separate phase, whose properties are orientation, atomic disordering, and chemistry dependency.[52,53]
The atomic interaction between solute atoms and Al plays an important role in affecting diffusion and GBS. However, it is important to recognize that the choice of atomic potentials may have a significant influence on the predicted rates of GBS and diffusion. For example, our simulations predict that Mg* inhibits GBS, whereas Namilae et al.[27] found that Mg slightly increased GBS tendency in Al. This discrepancy is caused by a difference between the potentials used in the two studies. Namilae et al.[27] used the EAM potential,[20] which predicts a negative heat of formation −0.036 eV/atom in Al12Mg7 phase and about 0.07 eV/atom for the dilute heat of solution of Mg in Al. The MEAM potential we used for Mg* predicts a heat of formation that is much more negative. Developing accurate alloy atomic potentials still remains a great challenge for MD simulations. Nevertheless, the general trend is that solutes with weak bonding with Al (such as Si) will accelerate GBS by increasing the mobility of Al atoms surrounding the solute atoms and, hence, increase the diffusivity of the GB. The heat of solution and the heat of formation can be used as a guide to the nature of the atomic interactions between the solute of atoms and Al. These values can be computed from more accurate ab initio calculations or can be measured from experiments. For example, the heat of solution of Si in Al is ~0.37 eV from first-principles calculations[54] and is ~0.51 eV from the experiment.[35] The computed heat of solution of Mg in Al is ~0.07 eV[38,53] and is in the 0.06 eV to 0.2 eV range from experiments.[35] Thus, both experimentally measured and computed heat of solution of Si is much larger than that of Mg in Al. The approach used here could be adopted as a general strategy for evaluating the influence of arbitrary solute atoms on GBS in Al. MD simulations with empirical potentials can be used to identify critical atomic scale parameters that influence the structure and energy of the GB and to explore the role of the parameters in controlling GBS resistance. The simulations can identify features of the potentials that lead to desirable behavior. Then, more accurate ab initio computations (such as DFT) can be used to identify solutes with relevant properties.
The reduction of threshold stress in low energy and the special low-Σ coincidence boundaries by Si addition greatly can influence the formability of polycrystalline Al alloys because low-Σ coincidence boundaries are common in these materials.[34,55,56] For recrystallized metallic materials, the percentage of special low-Σ coincidence boundaries can reach 50 pct in aluminum polycrystals with a grain size smaller than 30 μm. The materials of interest in the current work typically have a grain size of less than 10 microns. Because of the high resistance to GBS, these low-energy and low-Σ coincidence boundaries can be a limiting factor of the overall formability of the alloys during high-temperature forming. Cipoletti et al.[57] have shown how the transition from GBS mechanism to dislocation creep controlled high-temperature deformation varies with the percentage of sliding boundaries. Although MD simulations predict values of threshold stress for GBS that are significantly higher than macroscopic flow stresses observed experimentally during elevated temperature forming, the results are likely to show qualitatively that Si can reduce threshold stress. If we assume, for example, that adding Si decreases the amount of nonsliding boundaries from 43 pct to 23 pct in Cipoletti’s microstructure-based forming simulation, then the strain rate would increase from 1.2 × 10−3 to 2.2 × 10−3 seconds−1—an almost 100 pct increase—at 10 MPa, which translates to faster cycle times and lower cost forming.
4 Summary
Molecular dynamics simulations are used to investigate the effect of segregated Si and Mg* solute atoms in the GBs on GBS. One symmetric tilt GB and five asymmetric tilt GBs are used. The threshold stresses of GBS for these GBs with Si and Mg* solute atoms of two different concentrations are estimated and are compared with the threshold stress for the corresponding pure GBs. The results show that Si and Mg* solute atoms both have great influence on the sliding of Al GBs, but they operate by different mechanisms. Si impurities decrease the sliding threshold in high-energy GBs by approximately a factor of two by increasing the energy/mobility of surrounding Al atoms and enhancing GB diffusivity. Si impurities have less influence on low-energy GBs. Mg* impurities increase the sliding threshold of all GBs, independent of GB energy, by forming immobile clusters with surrounding Al atoms. We conclude that elements that have weaker bonding with Al, such as Si, will increase the mobility of neighboring Al atoms and so will increase the diffusivity of the GB region, enhancing GBS. Elements that have stronger bonding with Al, such as elements that form intermetallics with Al, will tend to form clusters with neighboring Al atoms and will decrease their mobility. This behavior will reduce the diffusivity of atoms in the GB region and will increase GBS threshold stress.
References
A.J. Barnes: Mater. Sci. Forum, 1999, vols. 304–06, pp. 785–96.
J.G. Schroth: Advances in Superplasticity and Superplastic Forming, Eds. E.M. Taleff, P.A. Friedman, P.E. Krajewski, R.S. Mishra, and J.G. Schroth, TMS, Warrendale, PA, 2004, pp. 9–20.
D. Wolf, V. Yamakov, S.R. Phillpot, A. Mukherjee, and H. Gleiter: Acta Mater., 2005, vol. 53, pp. 1.
R.N. Stevens: Phil. Mag., 1971, vol. 23, pp. 265–84.
R. Raj and M.F. Ashby: Metall. Trans., 1971, vol. 2, pp. 1113–27.
T.G. Langdon: Acta Metall. Mater., 1994, vol. 42, pp. 2437–43.
A. Ball and M.M. Hutchison: Met. Sci. J., 1969, vol. 3, pp. 1–6.
O.D. Sherby and J. Wadsworth: Prog. Mater. Sci., 1989, vol. 33, pp. 169–221.
R.C. Gifkins: J. Inst. Metals, 1967, vol. 95, pp. 373–77.
T.G. Nieh, J. Wadsworth, and O.D. Sherby: Superplasticity in Metals and Ceramics, Cambridge University Press, Cambridge, UK, 1997.
S. Agarwal, C.L. Briant, P.E. Krajewski, A.F. Bower, and E.M. Taleff: J. Mater. Eng. Perform., 2007, vol. 16, pp. 170–78.
A.F. Bower and E. Wininger: J. Mech. Phys. Solids, 2004, vol. 52, pp. 1289–317.
J.S. Vetrano, C.A. Lavender, and S.M. Bruemmer: Mater. Sci. Forum, 1994, vols. 170–72, pp. 225–30.
J.S. Vetrano, C.A. Lavender, and S.M. Bruemmer: Proc. 1995 124th TMS Annual Meeting, Las Vegas, NV, 1995, pp. 49–56.
J.S. Vetrano, C.H. Henager Jr., and V.Y. Guertsman: Advances in Superplasticity and Superplastic Forming, Eds. E.M. Taleff, P.A. Friedman, P.E. Krajewski, R.S. Mishra, and J.G. Schroth TMS, Warrendale, PA, 2004, pp. 225–32.
S.X. McFadden and A.K. Mukherjee: Mat. Sci. Eng. A, 2005, vol. 395, pp. 265–68.
X.T. Li, T.J. Li, X.M. Li, and J.Z. Jin: Ultrason. Sonochem., 2006, vol. 13, pp. 121–25.
T. Malis and M.C. Chaturvedi: J. Mater. Sci., 1982, vol. 17, pp. 1479–86.
X.Y. Liu and J.B. Adams: Acta Mater., 1998, vol. 46, pp. 3467–76.
X.Y. Liu, P.P. Ohotnicky, J.B. Adams, C.L. Rohrer, and R.W. Hyland: Surf. Sci., 1997, vol. 373, pp. 357–70.
X. Liu, X. Wang, J. Wang, and H. Zhang: J. Phys.: Condens. Matter, 2005, vol. 17, pp. 4301–08.
W.P. Green, M.A. Kulas, A. Niazi, K. Oishi, E.M. Taleff, P.E. Krajewski, and T.R. McNelley: Metall. Mater. Trans. A, 2006, vol. 37A, pp. 2727–38.
F. Weinberg: Trans. AIME, 1958, vol. 212, pp. 808–17.
M.D. Halliday and C.J. Beevers: J. Mater. Sci., 1971, vol. 6, pp. 1254–60.
M. Kato: Trans. Jpn. Inst. Met., 1969, vol. 10, pp. 215–22.
G. Mima, T. Oka, and T. Nishi: J. Jpn. Inst. Met., 1969, vol. 33, pp. 639–45.
S. Namilae, N. Chandra, and T.G. Nieh: Scripta Mater., 2002, vol. 46, pp. 49–54.
M.F. Ashby and R.A. Verrall: Acta. Metall., 1973, vol. 21, pp. 149–63.
J.H. Gittus: J. Eng. Mater., 1977, vol. 99, pp. 244–51.
H. Fukutomi and R. Horiuchi: J. Jpn. Inst. Met., 1983, vol. 47, pp. 457–61.
Y. Qi and P.E. Krajewski: Acta Mater., 2007, vol. 55, pp. 1555–63.
N. Du, A.F. Bower, P.E. Krajewski, and E.M. Taleff: Mater. Sci. Eng. A, 2008, vol. 494, pp. 86–91.
V.A. Ivanov and Y. Mishin: Phys. Rev. B, 2008, vol. 78, 064106.
T. Watanabe: Textures Microstructures, 1993, vol. 20, pp. 195.
J.R. Davies: ASM Specialty Handbook: Aluminum and Aluminum Alloys, ASM INTERNATIONAL, Materials Park, OH, 1993, pp. 579–80.
B.J. Lee, J.H. Shim, and M.I. Baskes: Phys. Rev. B, 2003, vol. 68, no. 14, 144112.
K. Gall, M.F. Horstemeyer, M. Van Schilfgaarde, and M.I. Baskes: J. Mech. Phys. Solids, 2000, vol. 48, pp. 2183–212.
B. Jelinek, J. Houze, S. Kim, M.F. Horstemeyer, M.I. Baskes, and S.G. Kim: Phys. Rev. B, 2007, vol. 75, 054106.
S. Nose: Mol. Phys., 2002, vol. 100, pp. 191–98.
S. Nose: J. Chem. Phys., 1984, vol. 81, pp. 511–19.
W.G. Hoover: Phys. Rev. A, 1985, vol. 31, pp. 1695.
J. Luo, V.K. Gupta, D.H. Yoon, and H.M. Meyer III: Appl. Phys. Lett., 2005, vol. 87, 231902.
V.K. Gupta, D.H. Yoon, H.M. Meyer III, and J. Luo: Acta Mater., 2007, vol 55, pp. 3131–42.
X. Shi and J. Luo: Appl. Phy. Lett., 2009, vol. 94, 251908.
A. Lindemann: Z. Phys., 1910, vol. 14, pp. 609.
Y. Qi, T. Cagin, W.L. Johnson, and W.A. Goddard III, J. Chem. Phys., 2001, vol. 115, pp. 385.
N. Du, Y. Qi, P.E. Krajewski, and A.F. Bower: Acta Mater., 2010, in press.
G.S. Painter and F.W. Averill: Phys. Rev. Lett., 1987, vol. 58, pp. 234–37.
J.R. Trelewicz and C.A. Schuh: Phys. Rev. B, 2009, vol. 79, 094112.
A.J. Detor and C.A. Schuh: Acta Mater., 2007, vol. 55, pp. 4221–32.
A.J. Detor and C.A. Schuh: Acta Mater., 2007, vol. 55, pp. 371–79.
S.J. Dillon, M. Tang, W.C. Carter, and M.P. Harmer: Acta. Mater., 2007, vol. 55, pp. 6208–18.
Y. Mishin, W.J. Boettinger, J.A. Warren, and G.B. McFadden: Acta Mater., 2009, vol. 57, pp. 3771–85.
N. Chetty, M. Weinert, T.S. Rahman, and J.W. Davenport: Phys. Rev. B, 1995, vol. 52, pp. 6313–26.
T. Watanabe and S. Tsurekawa: Acta Mater., 1999, vol. 47, pp. 4171–85.
T. Watanabe: Trans. Jpn. Inst. Met., 1986, vol. 27, pp. 73–82.
D.E. Cipoletti, A.F. Bower, Y. Qi, and P.E. Krajewski: Mater. Sci. Eng. A, 2009, vol. 504, pp. 175–82.
Acknowledgments
This work was supported through the General Motors/Brown Collaborative Research Laboratory on Computational Materials Science.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted December 14, 2009.
Rights and permissions
About this article
Cite this article
Du, N., Qi, Y., Krajewski, P.E. et al. The Effect of Solute Atoms on Aluminum Grain Boundary Sliding at Elevated Temperature. Metall Mater Trans A 42, 651–659 (2011). https://doi.org/10.1007/s11661-010-0326-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-010-0326-z