
Abstract
The biolistic method is reliable for delivering genes of interest into various species, but low transformation efficiency can be a limiting factor in its application. To test various conditions that could improve peanut transformation via particle bombardment, embryogenic tissues of the peanut cultivar Georgia Green were co-bombarded with two plasmids: one containing a green fluorescent protein gene and one containing a gene of interest plus a selectable marker. The fluorescence in bombarded embryogenic tissues was measured to evaluate transformation efficiency. A 4.6-fold improvement of transformation efficiency was achieved in stably transformed peanut lines by introducing protamine instead of conventional spermidine in a bombardment mixture with 70 ng/shot plasmid DNA and 50 μg/shot gold. Unexpectedly, the reduction of plasmid DNA from 700 to 70 ng/shot produced transgenic lines with significantly increased numbers of transgene copies. To determine the transgene copy number during plantlet regeneration, relative quantitative real-time polymerase chain reaction (qPCR) was established using fluorescently labeled universal library probes. A correlation of 95% was found for estimation of copy number between Southern blot and qPCR data. Given its speed and high-throughput nature, qPCR can be employed as an effective screening tool to separate high copy number events from low copy number events as early as the shoot formation stage of regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic transformation of peanut has been achieved by both biolistic and Agrobacterium-mediated transformation. A number of genes have been delivered into the peanut genome by co-cultivation with Agrobacterium tumefaciens (Li et al. 1997; Sharma and Anjaiah 2000; Rohini and Sankara Rao 2001; Anuradha et al. 2006; Bhatnagar-Mathur et al. 2007; Tiwari et al. 2008; Yang et al. 2011); however, only a limited number of peanut cultivars demonstrate relatively high transformation efficiency. Most of the elite peanut cultivars from the USA have not been transformed by Agrobacterium. Virulence of the bacterial strains and host–bacteria interactions may limit the success of Agrobacterium-mediated transformation (Gelvin 2003). Zygotic embryos, cotyledonary nodes, cotyledons, and leaves that can be regenerated by direct organogenesis are all used as target tissues for Agrobacterium-mediated transformation. The competency of these tissues for regeneration varies with genotype, and chimeric transgenic plants can be recovered from these differentiated tissues (McKently et al. 1995). On the other hand, biolistic transformation is a physical method for DNA introduction (Klein et al. 1987) and is less dependent on plant genotype, although embryogenic competence can vary among genotypes (Ozias-Akins et al. 1992). Due to the mode of proliferation of embryogenic tissue through the formation of secondary embryos, the recovery non-chimeric transgenic lines and germline transformants can be high with targeting of embryogenic tissues. Approximately 1–15% of bombarded peanut embryogenic clusters can produce transgenic lines (Ozias-Akins et al. 1993; Ozias-Akins and Gill 2001). Unfortunately, at least 1 yr is required to recover a transgenic plant through peanut tissue culture initiation, bombardment, and transgenic line regeneration. It is therefore of great interest to improve the transformation efficiency of bombardment.
Traditionally, bombardment involves coating microprojectiles (either gold or tungsten particles) with DNA in the presence of calcium chloride and spermidine (Klein et al. 1989). The addition of spermidine in the coating mixture stabilizes DNA structure and promotes the binding of DNA to gold particles (Klein et al. 1987; Thomas et al. 1996). Protamine, another cationic polyamine, protects coated DNA from DNase degradation longer than spermidine and improves transformation efficiency by 3-fold in rice (Sivamani et al. 2009). The amount of target DNA delivered per bombardment in peanut transformation ranges from 150 ng to 1 mg (Livingstone and Birch 1999; Magbanua et al. 2000; Yang et al. 2003; Athmaram et al. 2006). Reducing the amount of DNA coated on the microcarriers improved transformation efficiency and allowed recovery of more single-integration events in maize (Lowe et al. 2009).
Development of transgenic lines with stable and consistent levels of transgene expression is required for plant genetic engineering. Transgene expression is affected by many factors, including transgene copy number, integrity, orientation, and host genomic context of transgene loci (Iglesias et al. 1997). Particle bombardment often produces complex integration patterns, such as multiple copies, transgene rearrangements, and partial integration (Jackson et al. 2001). Simple transgene integration patterns (usually less than four copies) often resulted in more stable transgene expression or more consistent transgene-mediated host gene silencing by RNA interference constructs (Yang et al. 1998; Popelka et al. 2003; Altpeter et al. 2005; Livingstone et al. 2005; Chu et al. 2008b). Transgene expression is more consistent when the transgene cassettes are intact, arranged in a head-to-tail position, and preferably clustered in a gene-rich compartment of the host chromosome (Iglesias et al. 1997; Huang et al. 2002; Altpeter et al. 2005). Transgene silencing often occurs if the introduced DNAs are truncated and/or integrated in highly repetitive regions of the host genome (Matzke et al. 1994; Dorer and Henikoff 1997; Iglesias et al. 1997; Garrick et al. 1998). Transgenic events with high transgene copy numbers are undesirable since high transgene copy numbers have been associated with reduced transgene expression, instability over generations, and reduced seed development/viability (Vain et al. 2002). Determination of transgene copy number and elimination of transgenic lines with high copy number as early as the shoot regeneration stage would streamline the production of transgenic lines with high expression potential. Conventionally, Southern hybridization analysis is employed to estimate transgene copy number; it requires several grams of fresh tissue for DNA extraction (which is not feasible to obtain during the tissue culture stage) and is labor intensive and expensive. During transgene integration, if restriction sites are altered or the transgenes are arranged as concatamers, the number of hybridizing fragments will not be equal to the transgene copy number. Fiber-fluorescence in situ hybridization also has been used to determine the distribution and copy number of transgenes in the host genome (Jackson et al. 2001), but this requires training in cytogenetics and fluorescence microscopy and is unlikely to be widely adopted. Quantitative real-time polymerase chain reaction (qPCR) has been widely used for transgene copy number studies. qPCR is a closed-tube analysis, because the accumulation of PCR products is optically monitored at the end of each PCR cycle due to the presence of DNA-binding fluorescent molecules. The cross-point (Cp) value of the amplification curve is linearly correlated with the log of the initial concentration of a target gene in the sample, which allows for quantification of transgene copy number. qPCR has been used to determine zygosity of transgenic plants and transgene copy number in plants and animals (Ingham et al. 2001; Schmidt and Parrott 2001; Song et al. 2002; Prior et al. 2006; Joshi et al. 2008; Omar et al. 2008; Yi et al. 2008; Gadaleta et al. 2011). Previously, transgene zygosity in soybean and peanut was evaluated by real-time PCR (Schmidt and Parrott 2001), but transgene copy number has not been estimated by a real-time method in transgenic peanut. It was our first objective to bombard peanut embryogenic tissues with either protamine or spermidine in combination with varied levels of plasmid DNA to determine the conditions that improve peanut transformation efficiency. The second objective was to perform relative quantification of transgene copy number using an endogenous gene, actin depolymerizing factor (ADF), as an internal control. Feasibility of using real-time PCR to predict transgene copy number of 29 lines was tested and compared with Southern blot data.
Materials and Methods
Plasmid constructs.
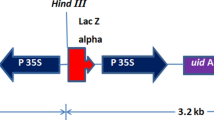
Two plasmids, p524EGFP.1 (Fleming et al. 2000) and pPPCH-V-Arah1_si (Fig. 1), were used for co-bombardment. In the Arah1_si plasmid, a 242-bp fragment of Ara h1, starting from position 1,242 bp of an Ara h1 complete mRNA sequence (GenBank accession L34402), was used to form an inverted repeat sandwiching a partial sequence for the β-glucuronidase (gus) gene (413 bp). The Ara h1 silencing construct was driven by a seed-specific promoter covering 660 to 1,546 bp of a Phaseolus vulgaris beta-type phaseolin storage protein gene (GenBank accession J01263), whose coding region starts from 1,547 bp (Fig. 1). A selectable marker gene for hygromycin resistance (potato ubiquitin-3 promoter, hph cds, nos terminator) was included in the Arah 1_si plasmid but not in the p524EGFP.1 construct.

Map of plasmid pPPCH-V-Arah1_si
Genetic transformation and plant regeneration.
Peanut embryogenic tissue cultures were initiated from mature zygotic embryos of the highly embryogenic peanut cultivar Georgia Green and subcultured on embryo maintenance medium (Ozias-Akins et al. 1993). Homogenous and translucent embryogenic tissues were recovered and used for bombardment after subculturing every 3–4 wk for 5 to 6 mo. After bombardment, all embryogenic tissues were kept in a dark incubator at 28°C until regeneration was initiated.
Plasmid DNA was isolated with the Qiagen Plasmid mini kit (Qiagen, Valencia, CA) and quantified with the Nanodrop 2000 (Thermo Scientific, Waltham, MA). Particle bombardment was performed using a PDS 1000/He apparatus (Bio-Rad, Hercules, CA). For coating microcarriers with DNA, either spermidine (Sigma, St. Louis, MO) or protamine (Sigma) was used with various amounts of 0.6-μm gold particles (Bio-Rad) and plasmid to determine the most suitable combination for peanut transformation (Table 1). Rupture and vacuum pressures were kept at 12,410 and 95 kPa, respectively. Briefly, six bombardment conditions were tested (Table 1), resulting from a combination of two levels of plasmid DNA (70 or 700 ng/shot; at both levels, the molar ratio of plasmids pPPCH-V-Arah1_si and p524EGFP.1 was kept at 1.5:1), two levels of gold (50 or 500 μg; remaining ingredients varied relative to gold concentration), and two coating agents for DNA binding (spermidine or protamine).
Embryogenic tissues were arranged in a 2.5-cm-diameter circle on a plate of maintenance medium and desiccated in a laminar flow hood for 2–3 h before bombardment. Six plates of tissue were bombarded for each experiment. Experiments P168 to P173 were performed on three consecutive days using 5-mo-old embryogenic tissues. Microcarrier preparation and bombardment conditions for P168, P169, and P171 were repeated 2 mo later in experiments P175, P176, and P177, respectively (Table 1). No statistical significance was found between the experiment dates; therefore, data were combined for analysis. Clusters of embryogenic tissue were spread across 8.5-cm plates 1 d after bombardment, and a number was assigned and tracked for each cluster during subculture. Four days post-bombardment, clusters were transferred to hygromycin (10 mg/L) selection on semi-solid medium for 2 wk and maintained on hygromycin-containing (20 mg/L) semi-solid medium for 4–5 mo. Proliferating clusters towards the end of selection were considered as putative transgenic lines.
Green fluorescent protein signal monitoring.
Expression of EGFP was evaluated by observation of tissues 1 and 4 mo after bombardment using a Zeiss SV11 epifluorescence stereomicroscope with a 480 ± 30-nm excitation filter and a 515-nm emission filter (Chroma Technology, Brattleboro, VT). At 1mo post-bombardment, three patterns of green fluorescent protein (GFP) fluorescence in tissues were observed (Fig. 2). In type I, the GFP signal was uniform throughout actively growing tissue. In type II, the GFP signal appeared in localized areas on actively growing tissue and showed chimeric transformation. In type III, the GFP signal was at single spots. After 4 mo, only type I GFP-positive clusters were counted. Since each bombardment condition was tested on six plates, and each plate was considered one biological replicate for that condition, GFP-positive signals of all types from one plate were summed and divided by the number of embryogenic clusters prior to bombardment. Data were analyzed by analysis of variance and means separated by the Tukey test using SAS software version 8.2 (SAS Institute, Inc, Cary, NC).

Three types of GFP signal observed 1 mo post-bombardment
Regeneration of transgenic plants.
Embryogenic hygromycin-resistant tissues were regenerated as previously described (Chu et al. 2008b). During the rooting stage, a small piece of leaflet from each plantlet was aseptically harvested for high-throughput DNA extraction (Xin et al. 2003). Primers for the hygromycin resistance gene cassette (sense: 5′-CCCCAATGTCAAGCACTTCCG-3′; antisense: 5′-CCGCGACGTCTGTCGAGAAG-3′) and/or phaseolin promoter region of the Arah 1_si construct (sense: 5′-GGCAATGTGTTTGTGTGTGGGTTG-3′; antisense: GCCGCTCCCATTTGACACTACG-3′) were used for PCR amplification to eliminate escapes from selection. The 642-bp amplicon of the phaseolin promoter region was also used as the probe for Southern blot. Amplification of the hygromycin resistance gene was carried out at 94°C for 5 min, followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s with a final extension at 72°C for 7 min and a 4°C soak. Amplification of the phaseolin promoter was the same as the above except that the annealing temperature was set at 58°C. Only PCR-positive plantlets were transferred to soil.
Southern blot analysis.
Genomic DNA was extracted from 2–3 g young leaf tissue of primary regenerants using a CTAB extraction procedure (Sharma et al. 2002). Genomic DNA (20 μg) was digested overnight with HindIII (New England Biolabs, Beverly, MA) to determine the transgene copy/insertion number. Within plasmid pPPCH-V-Arah1_si, HindIII has a single recognition site between the phaseolin promoter and the Ara h1 inverted repeat (Fig. 1). Digested DNA was separated on a 0.8% agarose gel and blotted onto Gene Screen Plus nylon membrane (NEN Research Products, Boston, MA). The phaseolin promoter probe was labeled with 32P-dCTP and the hybridization signal was detected with a Storm phosphor imager (Amersham Biosciences, Pittsburgh, PA).
Relative quantification of transgene copy number by qPCR.
The phaseolin promoter from the vector and the endogenous ADF sequences were submitted to the Universal Probe Library website (Roche Applied Science, Indianapolis, IN) for primer and probe design. A 78-bp fragment of the phaseolin promoter was amplified by primers (sense: 5′-TGAGGTGGCGGAAGAGAA-3′, antisense: 5′-CCCACACACAAACACATTGC-3′) and detected by HybProbe #9 (cat. No. 04685075001, Roche Applied Science). A 122-bp fragment of ADF was amplified by primers (sense: 5′-CATTGCTTGGTAATGATTCCATAA-3′, antisense: 5′-GGGACCTTCAAACAGAAGCA-3′) and detected by HybProbe #140 (cat. No. 04694244001, Roche Applied Science). The amplification of both products was performed on the Roche LC480 under PCR conditions of 95°C for 10 min (denaturation step) followed by 45 cycles of 95°C for 30 s and 55°C for 30 s. Fluorescent signal was captured at the end of each cycle. Both probes #9 and #140 were FAM labeled; therefore, amplification of each gene was performed in separate wells. qPCR was carried out in a total reaction volume of 10 μL containing 1× Roche Probe master and 0.5 μM of each primer. HybProbe #9 for the phaseolin promoter and HybProbe #140 for ADF were 0.5 and 0.1 μM, respectively. DNA samples were quantified with the Hoechst 33258 dye (Calbiochem, Darmstadt, Germany) and diluted to 0.78 μg/μL with 1× Tris-EDTA buffer (pH 8.0). Diluted genomic DNA (3.9 ng in 5 μL) was used for each reaction, and all samples were tested in triplicate. Twenty-nine transgenic lines with distinctive Southern blot banding patterns were included for qPCR study. The second derivative maximum method that identifies the Cp value at which the fluorescence curve turns sharply upward was used to perform advanced relative quantification of the transgene copy number (LightCycler 480 software, version 1.5.0, Roche Applied Bioscience).
Results and Discussion
The efficiency of bombardment was evaluated by analyzing GFP data collected at both 1 and 4 mo post-bombardment (Fig. 2). At 1 mo, three patterns of GFP signal were observed. Type I GFP signal was typified by uniform expression in actively growing tissue, which would be expected upon simultaneous integration of both GFP and hygromycin-resistance cassettes in cells undergoing secondary embryogenesis. Type II GFP signal, that is, GFP expression in large areas resulting in chimeric tissues, was probably produced from targeting a cell that was part of a preexisting embryo, resulting in the generation of partly transformed tissue, which will likely sort out following continued selection. Type III GFP signals were restricted to spots on the tissue resembling those observed in transient expression assays. It is possible that these cells were showing persistent transient expression or were stably transformed and expressing GFP but lacked co-integration of the hygromycin-resistance cassette; thus, they could no longer divide on the selection medium. By 4 mo, only type I GFP signal was observed, and overall GFP signal dropped 4-fold. The GFP signal counts at 4 mo post-bombardment could underestimate the total integration events. First, most of the cells in which GFP, but not the hygromycin-resistance gene, was integrated would be killed by hygromycin. Second, multiple GFP integration events in a cluster of tissue would be merged and become indistinguishable from one another after 4 mo of tissue culture. Regardless, all experiments were screened for GFP signal at the same time; therefore, comparison of the number of surviving GFP-positive clusters still could provide valuable information on the effect of bombardment conditions on the recovery of stably transformed events.
Significantly different transformation efficiencies based on GFP counts were found over the six bombardment conditions that had varied DNA, gold, and spermidine vs. protamine levels (Fig. 3). At 1 and 4 mo post-bombardment, experiments P168 and P175 showed the highest GFP expression frequency. Under this experimental condition, protamine was used at 170 ng/shot, gold at 50 μg/shot, and plasmid at 70 ng/shot (L/L/P). Previously, 700 ng to 1 μg DNA and 500 ng gold/shot in combination with spermidine was routinely used for bombardment experiments (Ozias-Akins et al. 1993; Singsit et al. 1997; Yang et al. 1998; Yang et al. 2003; Chu et al. 2008a, b). Reduction of DNA by at least ten times in the presence of protamine significantly increased the transformation efficiency over all the other conditions. Protamine was shown to protect DNA from degradation by endogenous cellular DNases longer than spermidine (Sivamani et al. 2009). With the same level of gold and plasmid DNA (compare bombardment conditions in L/L/P and L/L/S), addition of protamine significantly increased transformation efficiency by 15.2- and 4.1-fold at 1 and 4 mo, respectively. However, the inclusion of protamine was not the sole reason for the increase in transformation efficiency. When protamine was used with 700 ng DNA at 500 μg/shot gold levels (H/H/P), the transformation efficiency was not significantly different from those experiments with spermidine (H/H/S). Therefore, the combination of protamine with lower gold and plasmid levels elevated the transformation efficiency.

Transient expression and transformation efficiency measured by GFP signal counts divided by number of embryogenic clusters bombarded. a) Counted at 1 mo post-bombardment; b) Counted at 4 mo post-bombardment. For bar labeling, the first letter marks the level of plasmid being either low (L; 70 ng/shot) or high (H; 700 ng/shot); second letter marks the level of gold being either L (50 μg/shot) or H (500 μg/shot); third letter marks the presence of either protamine (P) or spermidine (S)
Of the approximately 700 plantlets regenerated on hygromycin-free medium after selection of embryogenic tissues for 4–5 mo on hygromycin-containing medium, 64% tested positive for the hph transgene by PCR. The release from hygromycin selection pressure during regeneration could have encouraged the growth of residual nontransformed cells. PCR genotyping for the presence of the transgene among regenerated plantlets eliminated escapes from the selection-free regeneration step. To further confirm transgene integration, Southern blot analysis for the phaseolin promoter in 40 primary regenerants that were PCR positive for hph showed that 38 (95%) contained the gene of interest (Fig. 4). This co-transformation frequency for linked genes is similar to previous observations in peanut (Wang et al. 1998). The number of hybridizing fragments on Southern blots varied from 1 to 16 among transgenic lines; patterns indicated that most lines represented independent transgene integration events (Table 2). Twenty-three lines had five or fewer copies, whereas eight lines had ten or more copies. Use of lower concentrations of cassette DNA for bombardment can reduce the complexity of integrations (Lowe et al. 2009). However, in our experiments, bombardments with 700 ng DNA produced transgenic lines with significantly fewer bands and less complex integration patterns than the experiments with 70 ng of DNA (P < 0.05). The lower level of DNA in our experiment is still more than double the published range that produces simpler transgene integration patterns. There may be a threshold of DNA amount in the bombardment mixture for simpler integration pattern that we have not yet approached. In addition, intact plasmids were used in our experiment instead of the expression cassette only. Linearized transgene constructs without vector backbone have been shown to reduce the complexity of transgene integration patterns (Fu et al. 2000).

Southern blot of genomic DNA from transgenic lines digested with HindIII and probed with a 642-bp amplified fragment from the phaseolin promoter sequence. The lighter band of the doublet in the plasmid control lane could come from incomplete digestion of the plasmid DNA
Using 30 independent transgenic lines, qPCR was conducted to determine transgene copy numbers. Previously, fluorophore-labeled TaqMan probes or nonspecific double stranded-DNA binding dyes such as Sybr green I were used for qPCR (Song et al. 2002; Omar et al. 2008). In our study, gene-specific amplicons were targeted for detection with universal library probes designed for transgene copy number estimation. Similar to a TaqMan probe, the universal library probe is a hydrolysis probe labeled with a FAM reporter at the 5′-end and a quencher at the 3′-end. Our preliminary data showed that the universal library probes gave specific amplification plots to both the phaseolin transgene and ADF endogenous control genes (data not shown).
Two methods were previously used to quantify transgene copy numbers; that is, absolute quantification by reference to a standard curve and relative quantification using a calibrator. Absolute quantification of transgene copy number relies on the accurate quantification of DNA and establishment of a standard curve (Livak and Schmittgen 2001; Bubner and Baldwin 2004). An alternative is to perform relative quantification by amplifying both the target transgene and an endogenous gene, whose copy number is constant relative to the host genome. In parallel, a transgenic line with known transgene copy number, preferably single copy, is chosen as the calibrator. Normalization of the target gene Cp value by the endogenous gene Cp value minimizes the effect of variation in template DNA concentration. The fold change of the normalized transgene Cp relative to that of a calibrator can be used to derive the copy number of transgenic lines. The relative quantification method is easier to perform than the absolute quantification method since it does not require a standard curve or precise quantification of DNA concentration. However, it does require that the transgene and the endogenous gene have similar amplification efficiencies (Livak and Schmittgen 2001). ADF was chosen as the reference gene. Separate reactions were carried out for endogenous genes and transgene, since the probes were both FAM labeled. Therefore, as long as the PCR efficiencies for both genes are similar, the transgene copy number can be evaluated.
One line that appeared to contain a single copy of the transgene based on Southern blot hybridization, 177-3-19, was used as the calibrator for copy number estimation. Quantification of transgene copy number requires the detection sensitivity of qPCR to be able to discern a 2-fold change of initial amount of DNA template. Amplification efficiencies of both the ADF-coding region and phaseolin promoter were determined from amplification plots of the calibrator line 177-3-19 following a 2-fold dilution of DNA between 3.9 and 125 ng. The amplification efficiencies for ADF and phaseolin promoter were 1.96 and 1.97, and the slopes were −3.42 and −3.39, respectively (Fig. 5). Ideally, if every cycle of PCR doubles the number of amplicons, the slope of the line should be −3.32 and the PCR efficiency should be 2. Our amplification plots for both genes suggest that the amplification conditions are close to optimum to perform transgene copy number quantification. The mean squared errors of the single data points fit to the regression line of ADF, and promoter plots were 0.00746 and 0.00503, respectively, indicating very little variation across the technical replications of each sample. Initially, 0.1 μM final concentration of HybProbe #9 for the phaseolin promoter was used, and the fluorescent signal peaked at 3.8, which was 4-fold lower than that of ADF, making it impossible to calculate the standard curve. Upon increasing the HybProbe #9 concentration to 0.5 μM, the amplification fluorescent signal was strong enough to produce a linear standard curve (Fig. 5). Subsequently, the quantification of the phaseolin promoter was detected using 0.5 μM final concentration of HybProbe #9.

qPCR Amplification plots generated by amplifying 2-fold dilutions of DNA extracted from calibrator line 177-3-19. a, Amplification plot of the endogenous ADF gene; b, amplification plot of the phaseolin promoter; c, standard curves of ADF and phaseolin promoter
The estimated transgene copy numbers ranged from 0.3 to 8. The correlation coefficient between Southern blot fragment number and transgene copy number determined by qPCR was 0.95 (Fig. 6), which is comparable to previous reports (Ingham et al. 2001; Song et al. 2002; Shou et al. 2004). Although both Southern blot and qPCR have been used to detect transgene copy numbers, the intrinsic differences between detection methods may not allow similar conclusions to be reached on determination of copy number (Bubner and Baldwin 2004). Southern blots detect the presence of a sequence by hybridizing a probe to restriction-digested DNA fragments. Incomplete digestion of DNA, alterations of restriction sites due to transgene integration/rearrangement, difficulty with inferring copy number in tandem repeats based on intensity differences, limitations for resolution of high molecular weight DNA fragments with standard agarose gel electrophoresis, and blotting efficiency would affect the accuracy of fragment number estimation. Particularly for the high copy number events, it is difficult to count the number of bands in a Southern blot image unambiguously. On the other hand, qPCR detects the presence of a very small portion of a transgene (usually less than 150 bp). Transgene rearrangement and truncation could make qPCR data misleading. In our study, the transgene copy number predicted by q-PCR is lower than that of Southern blot estimation. Regardless, the qPCR data are sufficient to distinguish high copy number events from low copy number events. This is a distinct advantage given that it usually takes 10–12 mo to regenerate transformed peanut plants and obtain progeny in the greenhouse. Eliminating high copy number lines at the regeneration stage saves considerable labor and space in the greenhouse and requires less than 1 d to process and evaluate 15 samples on a 96-well plate by qPCR. Further efficiency can be gained if the probes for the transgene and reference gene are labeled with different fluorescent dyes to allow multiplexing and testing of 30 samples per plate. This level of high throughput analysis supersedes the conventional Southern blot analysis, which takes at least 3–4 d for one round of hybridization.

Correlation of transgene copy numbers determined by Southern blot and qPCR
In conclusion, biolistic microcarrier preparation with protamine in combination with 70 ng/shot DNA and 50 μg/shot gold significantly improved the transformation efficiency of peanut embryogenic tissues. However, the pattern of transgene integration was more complex in experiments with 70 ng/shot DNA than with 700 ng/shot. The effect of bombardment conditions on transgene integration indicates the importance of optimizing bombardment conditions. The establishment of a qPCR protocol for estimating transgene copy number would simplify the screening of transgenic events as early as the shoot formation stage of regeneration.
References
Altpeter F, Baisakh N, Beachy R, Bock R, Capell T, Christou P, Daniell H, Datta K, Datta S, Dix PJ, Fauquet C, Huang N, Kohli A, Mooibroek H, Nicholson L, Nguyen TT, Nugent G, Raemakers K, Romano A, Somers DA, Stoger E, Taylor N, Visser R (2005) Particle bombardment and the genetic enhancement of crops: myths and realities. Mol Breed 15:305–327
Anuradha TS, Jami SK, Datla RS, Kirti PB (2006) Genetic transformation of peanut (Arachis hypogaea L.) using cotyledonary node as explant and a promoterless gus::nptII fusion gene based vector. J Biosci 31:235–246
Athmaram TN, Bali G, Devaiah KM (2006) Integration and expression of Bluetongue VP2 gene in somatic embryos of peanut through particle bombardment method. Vaccine 24:2994–3000
Bhatnagar-Mathur P, Devi MJ, Reddy DS, Lavanya M, Vadez V, Serraj R, Yamaguchi-Shinozaki K, Sharma KK (2007) Stress-inducible expression of At DREB1A in transgenic peanut (Arachis hypogaea L.) increases transpiration efficiency under water-limiting conditions. Plant Cell Rep 26:2071–2082
Bubner B, Baldwin IT (2004) Use of real-time PCR for determining copy number and zygosity in transgenic plants. Plant Cell Rep 23:263–271
Chu Y, Deng XY, Faustinelli P, Ozias-Akins P (2008a) Bcl-xL transformed peanut (Arachis hypogaea L.) exhibits paraquat tolerance. Plant Cell Rep 27:85–92
Chu Y, Faustinelli P, Ramos ML, Hajduch M, Thelen JJ, Maleki SJ, Ozias-Akins P (2008b) Reduction of IgE binding and non-promotion of Aspergillus flavus fungal growth by simultaneously silencing Ara h2 and Ara h6 in peanut. J Agric Food Chem 56:11225–11233
Dorer DR, Henikoff S (1997) Transgene repeat arrays interact with distant heterochromatin and cause silencing in cis and trans. Genetics 147:1181–1190
Fleming GH, Olivares-Fuster O, Fatta De-Bosco S, Grosser JW (2000) An alternative method for the genetic transformation of sweet orange. In Vitro Cell Dev Biol 36:450–455
Fu X, Duc LT, Fontana S, Bong BB, Tinjuangjun P, Sudhakar D, Twyma RM, Christou P, Kohli A (2000) Linear transgene constructs lacking vector backbone sequences generate low-copy-number transgenic plants with simple integration patterns. Transgenic Res 9:11–19
Gadaleta A, Giancaspro A, Cardone MF, Blanco A (2011) Real-time PCR for the detection of precise transgene copy number in durum wheat. Cell Mol Biol Lett 16:652–668
Garrick D, Fiering S, Martin DIK, Whitelaw E (1998) Repeat-induced gene silencing in mammals. Nat Genet 18:56–59
Gelvin SB (2003) Agrobacterium-mediated plant transformation: the biology behind the "gene-jockeying" tool. Microbiol Mol Biol Rev 67:16–37
Huang JM, Nandi S, Wu L, Yalda D, Bartley G, Rodriguez R, Lonnerdal B, Huang N (2002) Expression of natural antimicrobial human lysozyme in rice grains. Mol Breed 10:83–94
Iglesias VA, Moscone EA, Papp I, Neuhuber F, Michalowski S, Phelan T, Spiker S, Matzke M, Matzke AJ (1997) Molecular and cytogenetic analyses of stably and unstably expressed transgene loci in tobacco. Plant Cell 9:1251–1264
Ingham DJ, Beer S, Money S, Hansen G (2001) Quantitative real-time PCR assay for determining transgene copy number in transformed plants. Biotech 31(132–134):136–140
Jackson SA, Zhang P, Chen WP, Phillips RL, Friebe B, Muthukrishnan S, Gill BS (2001) High-resolution structural analysis of biolistic transgene integration into the genome of wheat. Theor Appl Genet 103:56–62
Joshi M, Keith Pittman H, Haisch C, Verbanac K (2008) Real-time PCR to determine transgene copy number and to quantitate the biolocalization of adoptively transferred cells from EGFP-transgenic mice. Biotech 45:247–258
Klein TM, Wolf ED, Wu R, Sanford JC (1987) High-velocity microprojectiles for delivering nucleic acids into living cells. Nature 327:70–73
Klein TM, Kornstein L, Sanford JC, Fromm ME (1989) Genetic transformation of maize cells by particle bombardment. Plant Physiol 91:440–444
Li Z, Jarret RL, Demski JW (1997) Engineered resistance to tomato spotted wilt virus in transgenic peanut expressing the viral nucleocapsid gene. Transgenic Res 6:297–305
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Method 25:402–408
Livingstone DM, Birch RG (1999) Efficient transformation and regeneration of diverse cultivars of peanut (Arachis hypogaea L.) by particle bombardment into embryogenic callus produced from mature seeds. Mol Breed 5:43–51
Livingstone DM, Hampton JL, Phipps PM, Grabau EA (2005) Enhancing resistance to Sclerotinia minor in peanut by expressing a barley oxalate oxidase gene. Plant Physiol 137:1354–1362
Lowe BA, Shiva Prakash N, Way M, Mann MT, Spencer TM, Boddupalli RS (2009) Enhanced single copy integration events in corn via particle bombardment using low quantities of DNA. Transgenic Res 18:831–840
Magbanua ZV, Wilde HD, Roberts JK, Chowdhury K, Abad J, Moyer JM, Wetzstein HY, Parrott WA (2000) Field resistance to tomato spotted wilt virus in transgenic peanut (Arachis hypogaea L.) expressing an antisense nucleocapsid gene sequence. Mol Breed 6:227–236
Matzke MA, Moscone EA, Park YD, Papp I, Oberkofler H, Neuhuber F, Matzke AJ (1994) Inheritance and expression of a transgene insert in an aneuploid tobacco line. Mol Gen Genet 245:471–485
McKently A, Moore G, Doostdar H, Niedz R (1995) Agrobacterium-mediated transformation of peanut (Arachis hypogaea L.) embryo axes and the development of transgenic plants. Plant Cell Rep 14:699–703
Omar AA, Dekkers MG, Graham JH, Grosser JW (2008) Estimation of transgene copy number in transformed citrus plants by quantitative multiplex real-time PCR. Biotechnol Prog 24:1241–1248
Ozias-Akins P, Gill R (2001) Progress in the development of tissue culture and transformation methods applicable to the production of transgenic peanut. Peanut Sci 28:123–131
Ozias-Akins P, Anderson WF, Holbrook CC (1992) Somatic embryogenesis in Arachis hypogaea L.: genotype comparison. Plant Sci 83:103–111
Ozias-Akins P, Schnall JA, Anderson WF, Singsit C, Clemente T, Adang MJ, Weissinger AK (1993) Regeneration of transgenic peanut plants from stably transformed embryogenic callus. Plant Sci 93:185–194
Popelka JC, Xu J, Altpeter F (2003) Generation of rye (Secale cereale L.) plants with low transgene copy number after biolistic gene transfer and production of instantly marker-free transgenic rye. Transgenic Res 12:587–596
Prior FA, Tackaberry ES, Aubin RA, Casley WL (2006) Accurate determination of zygosity in transgenic rice by real-time PCR does not require standard curves or efficiency correction. Transgenic Res 15:261–265
Rohini VK, Sankara Rao K (2001) Transformation of peanut (Arachis hypogaea L.) with tobacco chitinase gene: variable response of transformants to leaf spot disease. Plant Sci 160:889–898
Schmidt MA, Parrott WA (2001) Quantitative detection of transgenes in soybean [Glycine max (L.) Merrill] and peanut (Arachis hypogaea L.) by real-time polymerase chain reaction. Plant Cell Rep 20:422–428
Sharma KK, Anjaiah V (2000) An efficient method for the production of transgenic plants of peanut (Arachis hypogaea L.) through Agrobacterium tumefaciens-mediated genetic transformation. Plant Sci 159:7–19
Sharma AD, Gill PK, Singh P (2002) DNA isolation from dry and fresh samples of polysaccharide-rich plants. Plant Mol Biol Rep 20:415a–415f
Shou H, Frame BR, Whitham SA, Wang K (2004) Assessment of transgenic maize events produced by particle bombardment or Agrobacterium-mediated transformation. Mol Breed 13:201–208
Singsit C, Adang MJ, Lynch RL, Anderson WF, Wang AM, Cardineau G, Ozais-Akins P (1997) Expression of a Bacillus thuringiensis cryIA(c) gene in transgenic peanut plants and its efficacy against lesser cornstalk borer. Transgenic Res 6:169–176
Sivamani E, DeLong RK, Qu R (2009) Protamine-mediated DNA coating remarkably improves bombardment transformation efficiency in plant cells. Plant Cell Rep 28:213–221
Song P, Cai CQ, Skokut M, Kosegi BD, Petolino JF (2002) Quantitative real-time PCR as a screening tool for estimating transgene copy number in WHISKERS™-derived transgenic maize. Plant Cell Rep 20:948–954
Thomas TJ, Kulkarni GD, Greenfield NJ, Shirahata A, Thomas T (1996) Structural specificity effects of trivalent polyamine analogues on the stabilization and conformational plasticity of triplex DNA. Biochem J 319:591–599
Tiwari S, Mishra DK, Singh A, Singh PK, Tuli R (2008) Expression of a synthetic cry1EC gene for resistance against Spodoptera litura in transgenic peanut (Arachis hypogaea L.). Plant Cell Rep 27:1017–1025
Vain P, James A, Worland B, Snape W (2002) Transgene behaviour across two generations in a large random population of transgenic rice plants produced by particle bombardment. Theor Appl Genet 105:878–889
Wang A, Fan H, Singsit C, Ozias-Akins P (1998) Transformation of peanut with a soybean vspB promoter-uidA chimeric gene. I. Optimization of a transformation system and analysis of GUS expression in primary transgenic tissues and plants. Physiol Plant 102:38–48
Xin Z, Velten JP, Oliver ML, Burke JJ (2003) High-throughput DNA extraction method suitable for PCR. Biotech 34(820–824):826
Yang H, Singsit C, Wang A, Gonsalves D, Ozias-Akins P (1998) Transgenic peanut plants containing a nucleocapsid protein gene of tomato spotted wilt virus show divergent levels of gene expression. Plant Cell Rep 17:693–699
Yang H, Nairn J, Ozias-Akins P (2003) Transformation of peanut using a modified bacterial mercuric ion reductase gene driven by an actin promoter from Arabidopsis thaliana. J Plant Physiol 160:945–952
Yang CY, Chen SY, Duan GC (2011) Transgenic peanut (Arachis hypogaea L.) expressing the urease subunit B gene of Helicobacter pylori. Curr Microbiol 63:387–391
Yi CX, Zhang J, Chan KM, Liu XK, Hong Y (2008) Quantitative real-time PCR assay to detect transgene copy number in cotton (Gossypium hirsutum). Anal Biochem 375:150–152
Acknowledgments
The authors thank Evelyn Morgan for technical support, Joe Nairn, Warnell School of Forestry Resources, UGA for the phaseolin promoter construct, and reviewers and editors for their valuable revision suggestions. This research was supported by the Consortium for Plant Biotechnology Research and the Peanut Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Ted Klein
Rights and permissions
About this article
Cite this article
Chu, Y., Bhattacharya, A., Wu, C. et al. Improvement of peanut (Arachis hypogaea L.) transformation efficiency and determination of transgene copy number by relative quantitative real-time PCR. In Vitro Cell.Dev.Biol.-Plant 49, 266–275 (2013). https://doi.org/10.1007/s11627-013-9518-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-013-9518-8