
Abstract
Due to excessive proliferation and metastasis, glioma is the most common primary tumor in the central nervous system. Previous reports show simulated microgravity (SMG) has the ability to inhibit the proliferation and migration of cancer. The aim of this study was to evaluate the viability and migration of U251 cells in SMG environment. SMG induced apoptosis of U251 cells. The FAK/RhoA/Rock and FAK/Nek2 signaling events were attenuated by SMG to destabilize actin cytoskeleton and centrosome disjunction, which caused G2/M arrest and inhibition of cell viability and migration. Overexpressed FAK reversed SMG-induced inhibition of viability and migration in U251 cells, which increased downstream RhoA/Rock signaling and Nek2. These findings reveal novel pathways of FAK/RhoA/Rock and FAK/Nek2 are affected by SMG, and highlight an opportunity to expand therapeutic options in a variety of settings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioma is an aggressive cancer of the central nervous system with a 15-mo mean survival time and a dismal 5-yr survival rate of 5% (Ostrom et al. 2015). Despite standard care therapies, nearly all patients relapse. Due to genetic mutations and activation of pathophysiological signaling, glioma is excessive proliferation and metastasis (Dong et al. 2017). Thus, it is important to explore and understand the molecular mechanisms to improve therapeutic strategies.
As the most recognized member of the Rho GTPase family, RhoA activity drives the leading-edge polymerization of actin to form filamentous protuberances that regulate cell junctions, cytoskeleton formation, and cellular migration (Hetmanski et al. 2016). RhoA/Rock acts as an upstream regulator that activates the MAPK family, including p38, MAPK, and ERK cascades (Ghasemi et al. 2017). RhoA/Rock is a driver of tumor metastasis and invasion through MAPK, P53, or TGFβ signaling (Ghasemi et al. 2017). Nek2 is one of multiple cell cycle–regulated protein kinases that localize to the centrosome; it is required for mitotic progression and correct formation of the bipolar spindle (Bayliss et al. 2012). Nek2 has been reported to play a role in tumor progression, drug resistance, and tumorigenesis, and Nek2 overexpression is negatively correlated with survival of various malignancies (Liu et al. 2017). Several reports suggest RhoA/Rock and Nek2 are ideal therapeutic targets to fight metastasis and invasion of glioma (Yin et al. 2016; Liu et al. 2017).
Biological research of how space conditions, particularly zero gravity, impact the human body is becoming more common (Hughson et al. 2018). Given that spaceflight opportunities are rare and quite expensive, simulated microgravity (SMG) is a reliable and practical substitute (Aleshcheva et al. 2016). Increasing evidences indicate that the generation of ROS and oxidative stress is shown under SMG conditions and contributes to apoptosis initiation (Claro et al. 2014). According to Arun RP et al., PTEN/FOXO3/AKT axis is a main regulator of the cell fate under SMG conditions (Arun et al. 2017). Recently, SMG has been demonstrated to inhibit FAK, and FAK/RhoA-regulated mTORC1 and AMPK pathways reduce melanoma cell proliferation and metastasis (Tan et al. 2018). The development of malignancy is often concerned with abnormal FAK activity, such as oral squamous cell carcinoma and colon cancer cells (Li et al. 2018; Zhang et al. 2018). Moreover, class II histone deacetylase inhibitor suppresses cell migration and invasion in monomorphic malignant human glioma cells by inhibiting FAK/STAT3 signaling (Nam et al. 2017). However, the molecular mechanism of FAK pathways in SMG-induced changes of cell biology is still elusive, especially in glioma.
In this study, we investigated the effect of SMG on the viability and migration of U251 cells. We analyzed the potential molecular mechanism regulating the SMG-induced cellular responses by monitoring cell focal adhesions and associated signaling molecules, such as the FAK and RhoA/Rock signaling molecules, and Nek2 in cells under SMG. We found SMG inhibited FAK, reduced RhoA/Rock signaling and Nek2 expression, and attenuated glioma viability and migration. In addition, overexpressed FAK reversed SMG-induced inhibition of viability and migration in U251 cells, which increased downstream RhoA/Rock signaling and Nek2. Thus, FAK pathway is a crucial modulator in SMG-induced cell suppression in glioma.
Materials and Methods
Cell culture
U251 cells were obtained from the Cell Source Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, and cultured in DMEM medium (Gibco, Grand Island, NY) with 10% FBS (Gibco) at 37°C, 5% CO2.
Clinostat of SMG
Procedures were conducted according to previously described methods. Briefly, we followed Zhao et al. using SM-31 random locator (the Center for Space Science and Applied Research, Chinese Academy of Sciences; Beijing, China) (Zhao et al. 2016); cells were seeded in T25 culture flasks and cultured underground (1 g) overnight and flasks were placed at the center of the inner frame and rotated under SMG (10−3 g) at 37°C in a CO2 incubator with 30°/s angular velocity. The normal gravity control group was cultured without rotation.
Transient transfection
U251 cells were transfected with RhoA siRNA (Santa Cruz Biotechnology, Santa Cruz, CA; sc-29471), FAK siRNA (Santa, sc-29310), or FAK activation (Santa, sc-400,089) with Lipofectamine® 3000 (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol.
CCK8 assay
U251 cells were seeded at 3 × 103 cells per 96-well plate. Cell viability was assessed by CCK8 (Dojindo, Kumamoto, Japan). Briefly, after treatment, the 10 μl CCK8 solution was added to the culture medium, and the cultures were incubated for 1 h at 37°C, 5% CO2. The absorbance was measured at 450 nm using a microplate reader (Thermo Scientific, Carlsbad, CA).
Apoptosis analysis
Apoptosis of U251 cells was detected by Annexin-V-FITC Apoptosis Detection Kit (BD Biosciences, Franklin, NJ). The cells (1.0 × 105 cells/well) were seeded into six-well plates. Treated cells were washed twice with cold PBS and resuspended in buffer. The cells were treated according to the manufacturer’s instructions and measured with a flow cytometer (BD Accuri C6 Plus, BD Biosciences). After different treatments, the apoptotic cells were determined by the DeadEnd™ Colorimetric TUNEL System (Promega, Madison, WI) according to the manufacturer’s protocols. DAPI was used to stain cell nuclei. The stained sections were visualized with a fluorescence microscope (Nikon, Tokyo, Japan), and the TUNEL-positive cells were counted and analyzed using MetaMorph software.
Cell cycle analysis
For analysis of the cell cycle, the fixed cells were resuspended in PI/Triton-X staining solution containing 0.1% Triton-X, 50 μg/ml RNase A, and 50 μg/ml PI (Sigma, St. Louis, MO) for 30 min at 37°C in the dark, and subsequently analyzed by a flow cytometer, and the data were analyzed using the CellQuestk and ModFitk software.
SA-β-Gal staining
For analysis of the cell senescence, U251 cells with different treatments were trypsinized, fixed with 1% of formaldehyde for 15 min, and washed twice in PBS. Then, cells were stained with β-Gal Staining Kit (Sigma) and incubated at 37°C overnight in a dry incubator. Blue-stained cells and total cells were counted microscopically. β-Galactosidase expression = blue cells/total cells × 100%.
Wound healing assay
Cells were initially seeded on coverslips at a density of 5 × 104 /well. After the cells adhering to the coverslips for 6 h, a line was scratched with a sterile 200-μl pipette tip across the coverslips and the coverslips were inverted into the chamber. The wounded areas were photographed at the predicted times by inverted phase-contrast microscope (Nikon). The dot lines in pictures represent the healing of bilateral cells after scratch, the black lines represent the initial states, and the red lines represent the final states. At the initial states, the average width of an initial scratch is about 300 μm.
Transwell assay
Transwell assay was conducted with Transwell chamber (8 mm; Millipore, Billerica, MA). Matrigel Matrix (BD Biosciences) was diluted with DMEM at the ratio of 1:8 on ice, and 100 μl mixture was added into each chamber. Then, the chambers were kept at 37°C overnight. On the second day, 750 μl DMEM containing 10% FBS was added to the bottom of the chambers in the well and 250 μl cell suspension at the density of 1 × 105/ml was seeded into the top chamber. After a culture of 24 h, the non-invading cells on the top chamber were removed with cotton swab. Then, the chambers were washed twice with PBS, fixed with 75% alcohol for 10 min, stained with 0.5% Crystal Violet for 5 min at RT, and photographed in four independents × 10 fields/well. Cell invasion data was shown as the ratio of invasion cell number in each group compared with that in the control group.
Rho activation assay
RhoA activity was determined with a RhoA activation assay kit according to the manufacturer’s instructions (NewEast Biosciences, Malvern, PA). Briefly, cells were lysed in 500 μl of ice-cold 1× lysis buffer. Then, added 1 μl anti-active RhoA monoclonal antibody (NewEast Biosciences, 26904), thoroughly resuspend the protein A/G Agarose bead slurry by vortexing, quickly added 20 μL of resuspended bead slurry to each tube, incubated the tubes at 4°C for 1 h with gentle agitation, beads were washed, and proteins eluted in 20 μl of 2× reducing SDS-PAGE sample buffer by boiling for 5 min. Rho-GTP was measured by immunoblot analysis using anti-activated RhoA antibody (NewEast Biosciences, 21017).
Western blot assay
Western blot (WB) was conducted as described previously (Liu et al. 2018). Briefly, cells were lysed in lysis buffer containing proteinase inhibitor. Samples (30 μg protein per lane) were loaded on SDS-PAGE gels. After electrophoresis, the proteins were transferred to nitrocellulose membranes. Then, the membranes were blocked with 5% BSA and incubated at 4°C overnight with the appropriate primary antibodies. Immunoreactivity was detected by incubation with secondary antibodies (Abgent, Soochow, China; 1:20,000) followed by chemiluminescent substrate development (Thermo Scientific). Optical densities of the bands were calculated using a MiVnt image analysis system (Bio-Rad, Hercules, CA). The target proteins are normalized by the GAPDH, and the phosphorylation level is normalized by the total protein in the quantification of western blotting result. Primary antibodies are shown in Supplementary Table S1.
Immunofluorescence staining
Lamellar cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton-X for 10 min. They were blocked and incubated with primary antibody: Rock (1:100), p-LIMK2 (1:100), Nek2 (1:200), p-FAK (1:100), α-tubulin (1:200), and γ-tubulin (1:200), incubated with fluorescence-conjugated secondary antibodies for 1 h, at RT. Actin-Trakcer Green (C1033; Beyotime, Shanghai, China) was added at 1:100 dilution. Hoechest 33258 was used to stain cell nuclei. Images were captured on an inverted fluorescence microscope (IX71; Leica, Vitzla, Germany).
Statistical analysis
All data are presented as the mean ± SEM. Two-tailed Student’s t test or one-way ANOVA with Fisher’s post hoc test was used to determine significance. p < 0.05 was defined as statistically significant.
Results
SMG inhibited viability and migration of glioma cells
After SMG treatment, cell viability and migration of U251 cells were measured. We found SMG inhibited U251 cell viability in a time-dependent manner (Fig. 1a). In particular, the 48–96 h time points showed cell death with 45% decrease. Flow cytometry and TUNEL staining showed the SMG group had higher rate of death compared with control, at 72 h (Fig. 1b and c). More senescent cells were under SMG, approximately 30% (Fig. 1d). After SMG treated for 24 h, cell cycle analysis further showed SMG treatment significantly blocked the cell cycle in the G2/M phase (Fig. 1e). Through WB analysis, cleaved-caspase 3 and 9 were significantly upregulated, while Bcl2 and Bnip3 were downregulated under SMG treatment for 24 h, in U251 cells (Fig. 1f). In order to avoid the influence of cell motility from cell death, we evaluated the cell motility at SMG treated for 24 h. In wound healing assay, the scratched area was completely covered in the control group, compared with empty space with few migrated cells in the SMG group (Fig. 2a). Cell invasion data showed invasion capacity of the SMG group was reduced by 50% compared with the control group (Fig. 2b).

SMG induced U251 cell death. a CCK8 assay was used to analyze U251 cell proliferation in control vs. SMG conditions at differing time. After SMG treated for 72 h, cell apoptosis was detected by Annexin-V-FITC Apoptosis Detection Kit (b) and TUNEL staining (c), and metabolic activity was measured by β-Gal staining (d). e After SMG treated for 24 h, the fixed cells were resuspended in PI/Triton-X staining solution for 30 min at 37°C in the dark, and subsequently analyzed by a flow cytometer, and the data were analyzed using the CellQuestk and ModFitk software. f After SMG treated for 24 h, expressions of Bcl-2, Bnip3, and cleaved-caspase 3/9 were measured in U251 cells. Scale bars: 20 μm. Data are shown as the mean ± SEM, n = 5. *p < 0.05, ***p < 0.001, vs. control.

SMG suppressed U251 cell motor abilities. a In wound healing assay, the recovery of empty areas was used to assess the ability of cell migration after scratched with a sterile 200-μl pipette tip. Scale bars: 50 μm. b In Transwell assay, the non-invading cells on the top chamber were removed, the chambers were fixed and stained with 0.5% Crystal Violet, and then photographed in four independents × 10 fields/well. Cell invasion data was shown as the ratio of invasion cell number in each group compared with that in the control group. Scale bars: 20 μm. Data are shown as the mean ± SEM, n = 5. ***p < 0.001, vs. control.
SMG suppressed RhoA/Rock signaling and Nek2 expression in glioma cells
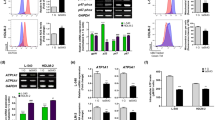
After SMG treatment, RhoA/Rock signaling molecules and Nek2 were detected in U251 cells. WB results showed the levels of pS188-RhoA, pY722-Rock2, pT508-LIMK2, and pS3-Cofilin, and Nek2 expression was suppressed after SMG treated for 24 h (Fig. 3a). pY722-Rock2, pT508-LIMK2, and Nek2 expressions were further verified by immunofluorescence staining (Fig. 3b). To confirm a role for RhoA/Rock signaling in glioma cells, we treated U251 cells with RhoA inhibitor Y-39983 (Selleck, S7935) and RhoA siRNA. Results from CCK8 assay, wound healing assay, and Transwell assay showed cell proliferation, migration, and invasion were reduced after RhoA suppression (Fig. 4a–c). In addition, WB results confirmed Rock/RhoA suppression significantly reduced the levels of pY722-Rock2, pT508-LIMK1, pT505-LIMK2, pThr18-MLC2, and pS3-Cofilin; Nek2 was not affected (Fig. 4d).

SMG alleviates RhoA/Rock signaling and FAK in glioma cells. U251 cells were cultured in SMG for 24 h, protein levels of RhoA/Rock signaling molecules and Nek2 were examined by WB (a), and immunofluorescence was used to detect phosphorylation levels of Rock2 and LIMK2, and Nek2 expression (b). c Integrin-β1, Integrin-β4, p-FAK, and FAK levels were detected after SMG treated for 24 h. Scale bars: 20 μm. Data are shown as the mean ± SEM, n = 5. *p < 0.05, vs. control.

RhoA/Rock signaling in glioma cells. After treated with RhoA inhibitor Y-39983 (1 μM) and RhoA siRNA, then viability and motor ability of U251 cells were analyzed by CCK8 assay (a, at 72 h), wound healing assay (b, at 24 h) and Transwell assay (c, at 24 h), and WB assay for RhoA/Rock pathway–related protein and Nek2 expression in U251 cells (d, at 24 h). Scale bars: b, 50 μm; c, 20 μm. Data are shown as the mean ± SEM, n = 5. **p < 0.01, ***p < 0.001, vs. control.
SMG suppressed cell viability and reorganized cytoskeleton by FAK inhibition
After SMG treatment, FAK signaling molecules and cytoskeleton were detected. In Fig. 3c, we found SMG significantly reduced FAK and pY397-FAK levels in U251 cells, and there was no significant difference of Integrin-β1 and Integrin-β4 in the control and SMG groups. To further verify the role of FAK, we also treated U251 cells with FAK inhibitor PF-573228 (Selleck, S2013) and FAK siRNA. Cell viability, migration, and invasion were decreased in the PF-573228 group, SMG group, and FAK-knockdown group (Fig. 5a–c, respectively). WB data showed SMG, PF-573228, and FAK siRNA suppressed FAK, pY397-FAK, pS188-RhoA, pY722-Rock, and Nek2 levels (Fig. 5d). Additionally, the analysis of the cellular concentration of RhoA-GTP in SMG-exposed glioma cells is included in Fig. 5e. RhoA activity was suppressed by SMG, PF-573228, and FAK siRNA, which was represented by GTP-RhoA expression in pull-down assay. As shown in IF results, SMG and PF-573228 suppressed FAK expression, but Y-39983 had no effect on FAK expression (Fig. 6a). Radial microtubule network became weak and lost membrane associated microfilament structures after cells were exposed to PF-573228, SMG, and Y-39983 (Fig. 6a). Immunofluorescence staining showed Nek2 and γ-tubulin in the PF-573228, SMG, and Y-39983 groups. PF-573228 and SMG decreased Nek2 expression; adding Y-39983 had no effect on Nek2 expression (Fig. 6b). PF-573228 and SMG suppressed centriole activity, shown by higher γ-tubulin green fluorescence intensity (Fig. 6b), and the distance between the two centrioles was reduced (Fig. 6c). There was no difference in the distance between the two centrioles in Y-39983 compared with the control group (Fig. 6c).

FAK in glioma cells. After treated with FAK inhibitor PF-573228 (1 μM), FAK siRNA, and SMG, then viability and motor ability of U251 cells were analyzed by CCK8 assay (a, at 72 h), wound healing assay (b, at 24 h) and Transwell assay (c, at 24 h), and WB assay for FAK pathway–related protein expression in U251 cells (d, at 24 h). RhoA activity was determined with a RhoA activation assay kit; the Rho-GTP was measured by immunoblot analysis using anti-activated RhoA antibody (e, after treated for 24 h). Scale bars: b, 50 μm; c, 20 μm. Data are shown as the mean ± SEM, n = 5. **p < 0.01, ***p < 0.001, vs. control.

SMG suppresses reorganized cytoskeleton. After treated with FAK inhibitor PF-573228 (1 μM), RhoA inhibitor Y-39983 (1 μM), and SMG for 24 h, cytoskeleton was stained with α-tubulin and Actin-Tracer Green, and FAK (a), and co-stained with Nek2 and γ-tubulin (b). Images were captured on an inverted fluorescence microscope (Leica, IX71). The distance between two centrosomes is measured through γ-tubulin staining, which shows the centrosomes in cells. Histogram analysis of distance between two centrosomes in U251 cells (c). Scale bars: 20 μm. Data are shown as the means ± SEM, n = 5. *p < 0.05, vs. control.
Overexpressed FAK reversed SMG inhibition
To verify the effect of FAK in SMG-induced U251 cell suppression, cells were treated with FAK activation and SMG. Results showed that FAK activation led to induction of pS188-RohA and higher Nek2 expression. FAK/RhoA and FAK/Nek2 pathways were reversed in FAK-activated cells, which suppressed under SMG (Fig. 7a). Results from pull-down assay showed RhoA activity was attenuated by SMG, and it was activated in the overexpressed FAK group (Fig. 7b). Results from CCK8, wound healing, and Transwell assays showed overexpressed FAK reversed SMG inhibition of cell proliferation, migration, and invasion in U251 cells (Fig. 7c–e, respectively).

Overexpressed FAK reverses SMG inhibition in glioma cells. U251 cells were transfected with FAK activation (Santa, sc-400089), and then treated with SMG or not. a Detected the level of FAK, p-FAK, p-RhoA, and Nek2 after SMG, FAK activation, and SMG + FAK activation treated for 24 h in U251 cells. b RhoA activity was determined by using a RhoA activation assay kit. U251 viability was detected by CCK8 assay at 72 h (c), the migration and invasion were analyzed by wound healing assay (d) and Transwell assay (e) at 24 h. Scale bars: d, 50 μm; e, 20 μm. Data are shown as the mean ± SEM, n = 5. **p < 0.01, ***p < 0.001, vs. SMG.
Discussion
SMG has been used to investigate the effects of microgravity and gravitation on various human biological events and specific functional modifications in ground experiments. SMG protects STO cells from undergoing apoptosis after exposure to moderate doses of radiation, possibly by decreasing the basal levels of caspase-3 activity, but vacuolated STO cells are induced by SMG alone (Beck et al. 2012). Such decrease in apoptosis could have dramatic consequences at the organismal level, by allowing accumulation of cells with persistent DNA damage and/or mutations (Risin and Pellis 2001). The cell death and growth arrest of cancer cells in simulated and actual microgravity are well documented in various cell types (Zhao et al. 2016). However, the specific mechanism of SMG on glioma proliferation and metastasis is not clear. In this work, we demonstrated SMG reduced RhoA/Rock and Nek2 pathways via FAK to suppress malignancy of glioma cells.
Our results showed a shift of the pathways involved in cell growth and motility when U251 cells were subjected to SMG. Some reports demonstrate that SMG affects cytoskeleton remodeling, as well as in cell cycle regulation and DNA damage response pathways in mouse fetal fibroblasts (Beck et al. 2014), FAK signaling in mesenchymal stem cells (Ratushnyy and Buravkova 2017), and FAK and Rhok signaling in the femoral artery of rat (Wang et al. 2015; Jiang et al. 2018). Recent evidences show FAK/RhoA pathway plays a crucial role in cancer cell proliferation and metastasis (McLean et al. 2005). For example, Procaine suppresses ERK/MAPK/FAK/RhoA pathways to inhibit proliferation and migration of colon cancer cells (Li et al. 2018). Tan et al. (2018) also show SMG significantly reduces FAK/RhoA signaling and mTORC1 kinase to alter cell proliferation and tumor metastasis in melanoma cell. In our study, we observed that the overall pathways of FAK/RhoA were altered: SMG suppresses FAK, GTP-RhoA, Rock, LIMK, MLC, and Cofilin activities, to recede cell proliferation and metastasis, but FAK overexpression was able to reverse the malignancy that inhibited by SMG in U251 cells (Fig. 8).

Schematic diagram of the signaling pathways affected by SMG in glioma cell via FAK activation.
Currently, the reduction in cell cycle during microgravity is mainly attributed to the disruption of the cytoskeletal elements resulting in G1 arrest (Papaseit et al. 2000). SMG arrests at G1 phase via PTEN/FOXO3/AKT pathway at 4 d in colorectal cancer cells (Arun et al. 2017). However, not all effect of SMG on blocking cell cycle is just G1 arrest. SMG causes C2C12 mouse muscle cells to decline muscle mass and retard their proliferation through accumulating at G2/M phase after 6-, 12-, and 24-h treatments (Benavides et al. 2013). Under SMG for 10 h, primary human IVD cells are accumulated in G2/M, reduced proliferation, and increased senescence (Franco-Obregon et al. 2018). We showed SMG arrested G2/M phase after 24-h treatment, this maybe SMG inhibited Nek2 to alleviate centrosome disjunction to prevent cell from entering mitosis and arrested G2/M phase. The accumulating evidences prove that Nek2 suppression regulates cell growth and proliferation via G2/M arrest (Lai et al. 2017). Involvement of Nek2 in microgravity is imminent, but less explored and understood. Additionally, we demonstrated PF-573228 and SMG suppressed Nek2 expression and decreased the distance between two centrioles, while Y-39983 had no adverse effect on these. Furthermore, FAK overexpression attenuated SMG-induced suppression of Nek2. These suggest that Nek2 reduction is independent of the RhoA/Rock pathway under SMG. Here, we verify SMG reduced FKA phosphorylation and downstream RhoA/Rock signaling and Nek2 activity. Thus, SMG reduces cytoskeleton stability and centrosome function via FAK/RhoA/Rock and FAK/Nek2 pathways.
In summary, this study demonstrates that FAK is a novel target of SMG in glioma. SMG arrests G2/M phase via FAK/Nek2 pathway and suppresses FAK/RhoA/Rock pathway to reduce glioma proliferation and migration. The FAK/RhoA/Rock and FAK/Nek2 signaling events are inactivated by SMG to destabilize actin cytoskeleton and centrosome disjunction. These data add to research and development of glioma therapeutics.
References
Aleshcheva G, Bauer J, Hemmersbach R, Slumstrup L, Wehland M, Infanger M, Grimm D (2016) Scaffold-free tissue formation under real and simulated microgravity conditions. Basic Clin Pharmacol Toxicol 119(Suppl 3):26–33
Arun RP, Sivanesan D, Vidyasekar P, Verma RS (2017) PTEN/FOXO3/AKT pathway regulates cell death and mediates morphogenetic differentiation of colorectal cancer cells under simulated microgravity. Sci Rep 7:5952
Bayliss R, Fry A, Haq T, Yeoh S (2012) On the molecular mechanisms of mitotic kinase activation. Open Biol 2:120136
Beck M, Tabury K, Moreels M, Jacquet P, Van Oostveldt P, De Vos WH, Baatout S (2012) Simulated microgravity decreases apoptosis in fetal fibroblasts. Int J Mol Med 30:309–313
Beck M, Moreels M, Quintens R, Abou-El-Ardat K, El-Saghire H, Tabury K, Michaux A, Janssen A, Neefs M, Van Oostveldt P, De Vos WH, Baatout S (2014) Chronic exposure to simulated space conditions predominantly affects cytoskeleton remodeling and oxidative stress response in mouse fetal fibroblasts. Int J Mol Med 34:606–615
Benavides DT, Franco-Obregon A, Egli M (2013) Gravitational force modulates G2/M phase exit in mechanically unloaded myoblasts. Cell Cycle 12:3001–3012
Claro S, Oshiro ME, Mortara RA, Paredes-Gamero EJ, Pereira GJ, Smaili SS, Ferreira AT (2014) gamma-Rays-generated ROS induce apoptosis via mitochondrial and cell cycle alteration in smooth muscle cells. Int J Radiat Biol 90:914–927
Dong Z, Lei Q, Yang R, Zhu S, Ke XX, Yang L, Cui H, Yi L (2017) Inhibition of neurotensin receptor 1 induces intrinsic apoptosis via let-7a-3p/Bcl-w axis in glioblastoma. Br J Cancer 116:1572–1584
Franco-Obregon A, Cambria E, Greutert H, Wernas T, Hitzl W, Egli M, Sekiguchi M, Boos N, Hausmann O, Ferguson SJ, Kobayashi H, Wuertz-Kozak K (2018) TRPC6 in simulated microgravity of intervertebral disc cells. Eur Spine J 27:2621–2630
Ghasemi A, Hashemy SI, Aghaei M, Panjehpour M (2017) RhoA/ROCK pathway mediates leptin-induced uPA expression to promote cell invasion in ovarian cancer cells. Cell Signal 32:104–114
Hetmanski JH, Schwartz JM, Caswell PT (2016) Modelling GTPase dynamics to understand RhoA-driven cancer cell invasion. Biochem Soc Trans 44:1695–1700
Hughson RL, Helm A, Durante M (2018) Heart in space: effect of the extraterrestrial environment on the cardiovascular system. Nat Rev Cardiol 15:167–180
Jiang M, Lyu Q, Bai YG, Liu H, Yang J, Cheng JH, Zheng M, Ma J (2018) Focal adhesions are involved in simulated-microgravity-induced basilar and femoral arterial remodeling in rats. Can J Physiol Pharmacol 96:772–782
Lai XB, Nie YQ, Huang HL, Li YF, Cao CY, Yang H, Shen B, Feng ZQ (2017) NIMA-related kinase 2 regulates hepatocellular carcinoma cell growth and proliferation. Oncol Lett 13:1587–1594
Li C, Gao S, Li X, Li C, Ma L (2018) Procaine inhibits the proliferation and migration of colon cancer cells through inactivation of the ERK/MAPK/FAK pathways by regulation of RhoA. Oncol Res 26:209–217
Liu H, Liu B, Hou X, Pang B, Guo P, Jiang W, Ding Q, Zhang R, Xin T, Guo H, Xu S, Pang Q (2017) Overexpression of NIMA-related kinase 2 is associated with poor prognoses in malignant glioma. J Neuro-Oncol 132:409–417
Liu W, Chen Y, Meng J, Wu M, Bi F, Chang C, Li H, Zhang L (2018) Ablation of caspase-1 protects against TBI-induced pyroptosis in vitro and in vivo. J Neuroinflammation 15:48
McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC (2005) The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer 5:505–515
Nam JH, Cho HJ, Kang H, Lee JY, Jung M, Chang YC, Kim K, Hoe HS (2017) A mercaptoacetamide-based class II histone deacetylase inhibitor suppresses cell migration and invasion in monomorphic malignant human glioma cells by inhibiting FAK/STAT3 signaling. J Cell Biochem 118:4672–4685
Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS (2015) CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro-Oncology 17(Suppl 4):iv1–iv62
Papaseit C, Pochon N, Tabony J (2000) Microtubule self-organization is gravity-dependent. Proc Natl Acad Sci U S A 97:8364–8368
Ratushnyy AY, Buravkova LB (2017) Expression of focal adhesion genes in mesenchymal stem cells under simulated microgravity. Dokl Biochem Biophys 477:354–356
Risin D, Pellis NR (2001) Modeled microgravity inhibits apoptosis in peripheral blood lymphocytes. In Vitro Cell Dev Biol Anim 37:66–72
Tan X, Xu A, Zhao T, Zhao Q, Zhang J, Fan C, Deng Y, Freywald A, Genth H, Xiang J (2018) Simulated microgravity inhibits cell focal adhesions leading to reduced melanoma cell proliferation and metastasis via FAK/RhoA-regulated mTORC1 and AMPK pathways. Sci Rep 8:3769
Wang ZC, Liu H, Bai YG, Yu JW, Zhang HJ, Cheng YP, Bao JX, Ren XL, Ma HZ, Ma J (2015) 28-Day hindlimb unweighting reduces expression of Rho kinase and inhibits its effects in femoral artery of rat. J Physiol Biochem 71:205–216
Yin M, Lu Q, Liu X, Wang T, Liu Y, Chen L (2016) Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ ROCK1 pathway. Biochem Biophys Res Commun 478:663–668
Zhang X, Feng H, Li Z, Guo J, Li M (2018) Aspirin is involved in the cell cycle arrest, apoptosis, cell migration, and invasion of oral squamous cell carcinoma. Int J Mol Sci 19. https://doi.org/10.3390/ijms19072029
Zhao T, Tang X, Umeshappa CS, Ma H, Gao H, Deng Y, Freywald A, Xiang J (2016) Simulated microgravity promotes cell apoptosis through suppressing Uev1A/TICAM/TRAF/NF-kappaB-regulated anti-apoptosis and p53/PCNA- and ATM/ATR-Chk1/2-controlled DNA-damage response pathways. J Cell Biochem 117:2138–2148
Acknowledgements
We thank Dr. Austin Cape at ASJ Editors for careful reading and feedback.
Funding
The present study was supported by the Youth Science and Technology Innovation Personnel Training Project of Shaanxi Province (No. 2017KJXX-84), the National Natural Science Foundation of China (Nos. 81402055 and 81602494), and the Natural Science Foundation of Shaanxi Province (No. 2016JM8014).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Editor: Tetsuji Okamoto
Banglian Deng, Rongrong Liu, and Xin Tian are the co-first authors to this article and contribute equally to this research project.
Electronic supplementary material
ESM 1
(DOCX 211 kb)
Rights and permissions
About this article

Cite this article
Deng, B., Liu, R., Tian, X. et al. Simulated microgravity inhibits the viability and migration of glioma via FAK/RhoA/Rock and FAK/Nek2 signaling. In Vitro Cell.Dev.Biol.-Animal 55, 260–271 (2019). https://doi.org/10.1007/s11626-019-00334-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-019-00334-7