
Abstract
Angiogenesis, or the formation of new blood vessels, is stimulated by angiogenic factors such as vascular endothelial growth factor (VEGF). Pigment epithelium-derived factor (PEDF) is a potent inhibitor of angiogenesis. To explore the mechanism by which PEDF acts, recombinant PEDF was expressed with a 6x-His tag (for purification) and a green fluorescent protein (GFP) tag. The PEDF fusion protein was confirmed to be active in inhibition of endothelial cell proliferation and migration. Direct binding of PEDF to both vascular endothelial growth factor receptor-1 (VEGFR-1) and VEGFR-2 was demonstrated in an in vitro assay similar to an enzyme-linked immunosorbent assay (ELISA). PEDF was shown by immune-confocal microscopy to be localized within treated endothelial cells. When VEGF-stimulated endothelial cells were incubated with PEDF the VEGF receptors showed intracellular localization. These data suggest that the interaction between PEDF and VEGFR-1 or VEGFR-2 may be a possible mechanism for inhibiting angiogenesis. PEDF may be binding to the VEGF receptors to promote their internalization and/or degradation to limit VEGF responses in treated cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tumor angiogenesis presents an important contribution to growth and development of solid tumors, providing access to nutrients and disposal of wastes without which tumors are unable to grow beyond a volume of approximately 0.4 mm3 (Gimbrone et al. 1972). On the balancing side, a variety of anti-angiogenic factors work to limit development of new vasculature (Hanahan and Folkman 1996). One of these anti-angiogenic factors is pigment epithelium-derived factor (PEDF), also known as early population doubling-level cDNA-1 (EPC-1) (Doggett et al. 1992; Dawson et al. 1999). PEDF loss is apparent in the neovascularization characteristic of diabetic retinopathy (Spranger et al. 2001). PEDF belongs to the serine protease inhibitors (serpin) superfamily, as a non-inhibitory member (Steele et al. 1993), which is susceptible to proteolytic cleavage by matrix metalloproteinases (Notari et al. 2005). A potent anti-angiogenic factor, PEDF induces apoptosis of endothelial cells, utilizing several distinct pathways (Chen et al. 2006; Ho et al. 2007, 2011; Volpert et al. 2002). Furthermore, PEDF blocks migration of endothelial cells in response to a wide variety of pro-angiogenic agents, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) among others (Dawson et al. 1999). Additionally, PEDF inhibits fibroblast growth factor-induced capillary morphogenesis by human and mouse endothelial cells in culture (Kanda et al. 2005). The variety of phenotypic effects induced by PEDF has led to increasing interest in pharmacological applications of PEDF (Becerra and Notario 2013; Chuderland et al. 2013; Craword et al. 2013; Orgaz et al. 2011).
Binding of cell surface molecules by PEDF has been investigated by several groups. A plasma membrane-linked phospholipase on the surface of retinal cells has been identified as a PEDF receptor (Notari et al. 2006; Subramanian et al. 2010, 2013). Binding of PEDF activates the enzyme, suggesting a cell signaling pathway that could promote retinal cell survival (Subramanian et al. 2010 2013). Phospholipase A2 engagement of PEDF on prostate cancer cells however, led to decreased production of IL8, key to prostate cancer progression (Hirsch et al. 2011). However, a variety of other surface molecules may recognize PEDF. A yeast two-hybrid assay discovered the non-integrin laminin receptor as a PEDF receptor whose engagement leads to inhibition of endothelial cell migration and to apoptosis (Bernard et al. 2009). A recent study found that this engagement leads to decreased fibronectin expression and subsequent downregulation of matrix metalloproteinases 2 and 9 (Hong et al. 2014). On bovine retinal endothelial cells, membrane-bound ATP synthase binds PEDF with high affinity, resulting in decreased production of extracellular ATP (Notari et al. 2010). Cell surface ATP synthase activity of human bladder carcinoma cells was also inhibited by PEDF leading to reduced tumor cell viability (Deshpande et al. 2012). PEDF has been shown to bind LRP6 to block activation of the wnt signaling pathway (Park et al. 2011). In addition, PEDF specifically binds extracellular matrix molecules such as hyaluronan (Becerra et al. 2008). This diverse set of effects leads ultimately to PEDF restriction of tumor growth and invasion.
The molecular mechanism by which PEDF exerts its anti-angiogenic effects is still unclear, however. Zhang et al. demonstrated a reciprocal transcriptional regulatory inhibition between VEGF and PEDF in retinal capillary endothelial cells; this was cell type specific: although PEDF decreased VEGF expression in Muller cells, VEGF did not negatively regulate PEDF (Zhang et al. 2006). Transcriptional downregulation of VEGF may be a result of decreased expression and nuclear import of Hif-1α, the major factor controlling VEGF expression (Zhang et al. 2011). Though predominantly viewed as a secreted protein, PEDF can be localized to the nucleus and has been shown to be imported by transportin SR2 in cells expressing ectopic PEDF (Anguissola et al. 2011). In addition, PEDF blocked VEGF binding to VEGF receptor 2 (VEGFR-2) in vitro and on the surface of retinal capillary endothelial cells (Zhang et al. 2006). PEDF-induced activation of γ-secretase leads to intramembrane proteolysis of VEGF receptors 1 and 2 and internalization of the C-terminal domain of VEGFR-1 (Cai et al. 2006; Ablonczy et al. 2009). As the binding of PEDF to various cell surface moieties generates different effects, the anti-tumorigenic activity of PEDF can be characterized as multi-modal (Becerra and Notario 2013).
To further understand the effect of PEDF on VEGF-mediated angiogenesis, we generated and purified a GFP-PEDF fusion protein that maintains anti-proliferative and anti-invasive activity on human umbilical vein endothelial cells. The fusion protein directly binds both VEGFR-1 and VEGFR-2 in an in vitro assay. Both VEGF receptors and PEDF are localized internally within endothelial cells after PEDF treatment. These data support a direct interaction between PEDF and the VEGF receptors as a mechanism for inhibition of angiogenesis.
Materials and Methods
Protein generation.
Human PEDF containing a C-terminus six histidine tag was engineered into pEGFP-N1 mammalian expression vector (BD Biosciences, San Jose, CA). The recombinant PEDF-6x-His-GFP fusion protein was expressed transiently in HEK293 cells. Cells were seeded at 4 × 105 cells/ml in a cell factory containing 1 l of medium consisting of DMEM, 10% fetal bovine serum, and 250 mg geneticin (Invitrogen, Grand Island, NY), transfected with the PEDF-6x-His-GFP DNA construct using Lipofectamine™ 2000 Transfection Reagent (Invitrogen), and grown in 293 SFMII serum-free media (Invitrogen) with protease inhibitors (Sigma, St. Louis, MO). The medium was collected, and all subsequent steps were carried out at 4°C. The secreted recombinant PEDF-6x-His-GFP fusion protein was harvested by centrifugation and filtered using a 0.8-μm sterile filter. The recombinant PEDF-6x-His-GFP fusion protein was then purified using Talon affinity chromatography (Clontech, Mountain View, CA). The purified PEDF-6x-His-GFP fusion protein was dialyzed against phosphate buffered saline with protease inhibitors at neutral pH, and then concentrated using an Amicon Ultra 30 kDa MWCO centrifuge concentrator. The PEDF-6x-His-GFP fusion protein was analyzed by absorbance at 280 nm, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblot to determine protein concentration, yield, and purity. PEDF was detected with mouse anti-human PEDF diluted at 1:500 in Tris-buffered saline and Tween 20 (TBST; R&D systems, Minneapolis, MN) and horseradish peroxidase conjugated anti-mouse diluted at 1:10,000 in TBST. To detect the 6x-His tag, anti-HIS-HRP (Clontech) antibody was used at a 1:10,000 dilution. To detect the GFP tag, anti-GFP-HRP (Invitrogen) was used at a 1:10,000 dilution.
Endothelial cell migration assay.
Biological activity of the purified PEDF-6x-His-GFP was tested in a modified Boyden chamber migration assay using the Chemicon Cell Invasion Assay kit (Millipore, Billerica, MA) and VEGF as the chemoattractant. Human umbilical vein endothelial cells (HUVEC, Lonza No. C2517A, Basel, Switzerland) were cultured with complete growth media (Lonza, Basel, Switzerland), harvested at passage 6, and resuspended at 2 × 105 cells/ml in serum free medium (Lonza) with 0.01% BSA. Cells (2 × 104/well) were plated in the upper chamber of a 96-well format insert. Recombinant VEGF162 (R&D Systems) was added at 10 ng/ml to the bottom chamber in serum-free media to serve as chemoattractant. PEDF-6x-His-GFP fusion protein was added at 1 ng/ml to the upper chamber. After migration through the membrane, the viable cells were removed using a detachment buffer and subsequently detected using CyQuant GR dye (Molecular Probes, Grand Island, NY).
Endothelial cell viability assay.
To further evaluate the activity of PEDF-6x-His-GFP fusion protein, a cell viability assay was performed to determine if recombinant PEDF could inhibit proliferation of VEGF-stimulated endothelial cells. HUVECs were plated at 1.6 × 104 cells/well in 96-well white clear-bottom plates (NUNC, Rochester, NY). PEDF-6x-His-GFP fusion protein was added at 100 μg/ml and incubated at 37°C for 30 min prior to the addition of 10 ng/ml of VEGF162 (R&D Systems). The cells were incubated at 37°C for 24 h before measuring cell viability. Cell viability was measured using Cell Titer Glo reagent (Promega, Madison, WI) to detect metabolically active cells.
Binding ELISA: protein-protein Interactions.
VEGF receptor fusion proteins (VEGFR-1: R&D Systems No. 321-FL and VEGFR-2: R&D Systems No. 357-KD) containing a human IgG Fc region and 6x-histidine tag were used for the binding enzyme-linked immunosorbent assay (ELISA); 96-well Maxisorb clear plates (NUNC) were coated with 50 μl of 2 μg /ml goat anti-human IgG, Fcγ fragment specific (Jackson Immunoresearch, West Grove, PA) in bicarbonate buffer at pH 9.4 (Pierce) overnight. The plates were blocked using Starting block TBS buffer (Pierce) for 1 h at room temperature, and then washed three times with 300 μl of wash buffer (PBS; 0.01% Tween-20); 50 μl of 2 μg/ml VEGFR-1 or VEGFR-2 in assay buffer (PBS, 0.01% Tween-20) was added to the plate and incubated for 1 h shaking at room temperature. The microtiter plate was washed, and then 50 μl of PEDF-6x-His-GFP fusion protein at various concentrations starting at 5 μg/ml with subsequent 1:2 dilutions in assay buffer (PBS, 0.01% Tween-20) was added to the plate and incubated for 2 h shaking at room temperature. A non-VEGF receptor-binding protein (Toll-like receptor family ECD his-tagged protein) (Duffy et al. 2007) was used as a negative control. The microtiter plate was washed, and then 50 μl of mouse anti-human PEDF (R&D Systems No. MAB1177) at 1 μg/ml was added and incubated for 1 h shaking at room temperature. The microtiter plate was washed, and then 50 μl of goat anti-mouse HRP-conjugated (Jackson Immunoresearch) at 1 μg/ml was added and incubated for 1 h shaking at room temperature. The microtiter plate was washed, and then binding was detected using TMB substrate (EMD Biosciences, Billerica, MA). The reaction was stopped using 0.2 N sulfuric acid and measured by absorbance at OD450 nm. The assay was performed twice with triplicate wells per assay. The data are presented as means +/- standard error of the mean (n = 3) and the averaged background, or zero, value was subtracted for each data set.
Localization assays using fluorescent microscopy.
HUVECs (Lonza) were plated on cover slips in 24-well plates at 1 × 105 cells/ml in 1 ml of serum-free media (Lonza) and grown overnight at 37°C. Recombinant PEDF-6x-His-GFP fusion protein was added at 10 μg/ml alone or with 25 ng/ml VEGF165 (R&D Systems No. 293-VE) and incubated for 1, 2, 4, 8, and 24 h at 37°C. At each time point, the cells were washed with 1× PBS, fixed with 4% paraformaldehyde in 1× PBS for 30 min at 37°C, permeabilized with 0.1% TritonX-100 in 1xPBS for 15 min, incubated with signal enhancer (Invitrogen) for 30 min, and blocked with Blotto blocking buffer (Sigma) for 30 min. The cells were then fluorescently stained or probed with primary and secondary antibodies. The nuclei were stained with a fluorescent probe, 4′,6-diamidino-2-phenylindole (DAPI; Sigma), the actin filaments were stained with Phalloidin Alexa Fluor 647 (Invitrogen), and the PEDF-6x-His-GFP fusion protein was probed with anti-GFP Alexa Fluor 488 (Invitrogen). VEGF receptors were probed with primary antibodies (goat anti-VEGFR-1 and mouse anti-VEGFR-2—R&D systems) and stained with secondary antibodies (anti-goat Alexa Fluor 568 (VEGFR-1) and anti-mouse Alexa Fluor (VEGFR2), Invitrogen. After the cells on the cover slips were stained, the cover slips were mounted and sealed onto glass slides using mounting media (EMS) and nail polish (EMS). The slides were visualized using confocal microscopy (Perkin Elmer) at wavelengths of 405 nm (DAPI), 647 nm (Phalloidin), or 488 nm (GFP) to see multiple optical sections of the cells. The images obtained were analyzed using Perkin Elmer Imaging software.
Results
Recombinant protein generation.
Human PEDF containing a C-terminus six histidine tag was engineered into pEGFP-N1 mammalian expression vector (Fig. 1a ), expressed transiently in HEK293 cells, and purified using immobilized metal affinity chromatography (IMAC). The purified protein product was assessed by SDS-PAGE and immunoblots (Fig. 1b ). The PEDF-6x-His-GFP fusion protein calculated molecular weight should be 75 kDa, but the SDS-PAGE results show protein present at approximately 90 kDa, possibly due to post-translational modification, and another band resolving at 50 kDa (Fig. 1b (A)). N-linked glycosylation was also observed by Duh et al. (2002) for recombinant PEDF expressed in human embryonic kidney cells. The anti-His, anti-PEDF, and anti-GFP Western blots all show protein products at approximately 90 kDa (full-length) and at 50 kDa (Fig. 1b (B, C)). The anti-GFP Western blot also showed a lower molecular weight cleavage product around 30 kDa in the input and unbound material (Fig. 1b (D, E)) but not in the final purified product (F), since the protein was concentrated using a 30-kDa MWCO membrane. As the smaller band was not detected with anti-PEDF or anti-His antibodies, the results suggest that the fusion protein may be cleaved just before the C-terminal GFP tag.

(a) Protein construct design. The full-length expressed protein contains a C-terminal 6x-histidine and GFP tag for a total predicted molecular weight of 75 kDa. PEDF-HIS is 50 kDa, and the GFP tag is 25 kDa. (b) Analysis of purified protein by SDS-PAGE and western blot. Full-length PEDF-6x-His-GFP (~90 kDa) and a cleavage product at 50 kDa are shown here by SDS-PAGE (A) and western blots (B–F). The anti-GFP western also shows the 30-kDa cleavage product in the input (D) and unbound fraction (E) but not in the purified product (F). The PEDF fusion protein is cleaved just before the GFP tag and the lower molecular weight product is removed by centrifugal concentration of the protein (F). The low molecular weight band was not present on blots detected by anti-PEDF or anti-His.
Endothelial cell migration and viability assays.
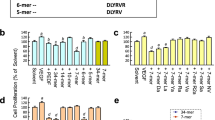
To determine if the PEDF-6x-His-GFP fusion protein was active as a functional angiogenic inhibitor, it was tested in a modified Boyden chamber migration assay using VEGF as the chemoattractant. HUVECs that are stimulated with VEGF to induce endothelial cell migration can be inhibited by PEDF (Fig. 2a ). A significant increase in cell migration is observed with 10 ng/ml of VEGF stimulation. When the PEDF fusion protein is added to the cells at 1 ng/ml, we see a significant decrease of endothelial cell migration toward VEGF (Fig. 2a ). Addition of the PEDF to cells directly may permit the inhibitor to block the receptor, preventing migration through the barrier. This possibility was tested in vitro in further experiments (see Fig. 3).

(a) Endothelial cell migration assay HUVECs were plated in Boyden chamber inserts at 2 × 105 cells/ml in serum-free media. VEGF was added in the bottom chamber at 10 ng/ml in serum-free media to serve as chemoattractant. PEDF-6x-His-GFP fusion protein was added to cells at 1 ng/ml and incubated 24 h at 37°C. The migrated cells were lysed from the membrane, and the viable cells were measured using a fluorescent dye to quantitate cell migration. Relative fluorescence units were measured to represent the number of migrated cells. The data are presented as means ± standard error of the mean (n = 2). (b) Endothelial cell viability assay. HUVECs cultured in complete growth media were plated in 96-well plates at 2 × 105 cells/ml in serum-free media. PEDF-6x-His-GFP fusion protein was added to cells at 100 μg/ml and incubated at 37°C for 30 min before adding 10 ng/ml VEGF162. The cells were incubated at 37°C for 24 h, and then cell viability was measured by Cell Titer Glo reagent. Relative luminescence units were measured to represent cell number. An increase in cell viability is interpreted to be a result of proliferation. The data are presented as means ± standard error of the mean (n = 4). Statistical analysis was done using a Student’s t test for comparisons. A value of p < 0.05 was considered statistically significant. ***p < 0.001 + VEGF vs. −VEGF; ***p < 0.001 + VEGF vs. +VEGF and PEDF.

Binding of the VEGFR-1 (a) and VEGFR-2 (b) to PEDF-6x-His-GFP fusion protein by ELISA. The assay was performed by coating the plate with goat anti-human Fc, binding the VEGFRs at 2 μg/ml, adding PEDF-6x-His-GFP fusion protein and control (Toll-like receptor ECD-HIS) at decreasing concentrations starting at 5 μg/ml, and then detecting with primary and secondary HRP labeled antibodies. The data are presented as means ± standard error of the mean (n = 3). OD of the HRP product was measured at 450 nm.
To further evaluate the activity of PEDF-6x-His-GFP fusion protein, a cell viability assay was performed to determine if recombinant PEDF could inhibit VEGF-stimulated cell proliferation. Addition of VEGF (10 ng/ml) to culture medium led to a strong increase in cell viability, presumably due to increased proliferation. PEDF-6x-His-GFP fusion protein does inhibit VEGF-induced cell proliferation at 100 μg/ml (Fig. 2b ). At lower PEDF concentrations, only partial inhibition of VEGF stimulation was observed (data not shown). The high concentration required to demonstrate inhibition of proliferation (100 μg/ml) is comparable with 3 μM for a 75-kDa protein. Subramanian et al. detected maximal PEDF inhibition of tumor cell viability at 100 nM, however in the absence of VEGF (Subramanian et al. 2012). Duh et al. (2002) were able to demonstrate inhibition of VEGF-induced viability of bovine retinal epithelial cells using 100 ng/ml PEDF. Size exclusion HPLC of our purified recombinant PEDF showed that a significant fraction was aggregated; it is possible that aggregation reduced the ability of the protein to induce proliferation of HUVECs. These results confirm that the PEDF-6x-His-GFP fusion protein is active and functional by inhibiting endothelial cell migration and proliferation.
Binding ELISA: PEDF binding to VEGF receptors.
A possible mechanism by which PEDF may be inhibiting angiogenesis is through directly engaging the VEGF receptors. We investigated the binding of the VEGF receptors to PEDF by ELISA to determine if PEDF is binding and interacting with the VEGFR-1 and/or VEGFR-2. The results (Fig. 3a, b ) showed that PEDF binds strongly to VEGFR-1 and weakly to VEGFR-2. The difference in binding affinity between the two receptors was approximately 10-fold; while PEDF exhibited 50% binding to VEGFR-1 at 0.16 μg/ml, 50% binding to VEGFR-2 was only achieved at a concentration of 2.0 μg/ml. A His-tagged protein (Toll-like receptor ECD) (Duffy et al. 2007) not cognate for the VEGF receptors was used as a negative control in this assay and did not bind to either of the VEGF receptors. A previous study had demonstrated that PEDF prevented 50% binding of VEGF to the VEGFR-2 receptor in vitro at 10 nM (Zhang et al. 2006), which for our construct would be approximately 0.75 μg/ml, in reasonable agreement given the difference in format. A direct interaction was not tested by these researchers, and the inhibition could be explained by PEDF binding to either VEGF or VEGF receptors (Zhang et al. 2006). Therefore, binding of PEDF directly to VEGFR-1 and VEGFR-2 in vitro presents a reasonable mechanism by which PEDF blocks VEGF-induced events.
Localization of PEDF.
To determine if PEDF remains on the surface of endothelial cells or if it becomes internalized, endothelial cells were treated with PEDF alone or with PEDF and VEGF and monitored by confocal microscopy over 24 h. The results showed that the PEDF-6x-His-GFP fusion protein localizes inside the endothelial cell with and without VEGF stimulation over a 24-h time course (Fig. 4a (A–H)). One hour after addition, PEDF is widely and fairly evenly distributed, presumably on the cell surface but is more evident in perinuclear regions by 2 h. It is possible that some PEDF is taken up and targeted to lysosomes. Actin stress fibers are more prominent in cells prior to treatment (Fig. 4b (A, D)) than 1–2-h post-PEDF but are once again well defined at 24 h time points (Fig. 4a (D, H)). Note the change in cell shape from untreated cells (Fig. 4b (A, D)) to a more compact form at 1–2 h after treatment. By 24 h after treatment (Fig. 4a (D, H)), cells appear once more to be flattened and extended. Cells treated with VEGF as well as PEDF do show some co-localization of PEDF and DAPI (nuclear) staining at 8 h (Fig. 4a (G)). The localization of the VEGFRs was also monitored. The results showed that VEGFR-1 and VEGFR-2 are widely distributed and somewhat punctate without PEDF stimulation, and when endothelial cells are incubated with the PEDF-6x-His-GFP fusion protein, the detection of VEGFR-1 and VEGFR-2 is concentrated in perinuclear regions with or without VEGF (Fig. 4b (A–F)). The total intensity of VEGFR appeared to be lower, particularly the dispersed forms, in cells treated with PEDF. The perinuclear localization may represent largely newly synthesized VEGFRs, as the appearance is consistent with localization within the secretory pathway. Some VEGFRs are localized apparently internal to the plasma membrane after PEDF treatment (Fig. 4b (E)), consistent with endocytic uptake and potential degradation as suggested by other investigators (Orgaz et al. 2011; Chuderland et al. 2013).

(a) PEDF localizes inside HUVECs. HUVECs were plated at 1 × 105 cells/ml in serum free media and incubated overnight at 37°C. Recombinant PEDF-6x-His-GFP fusion protein was added at 10 μg/ml alone (A–D) or with 25 ng/ml VEGF (E–H) and incubated for 1, 2, 4 (not shown), 8, and 24 h at 37 °C. The cells were fixed and stained at each time point with fluorescent probes: DAPI (nuclei, blue), Phalloidin Alexa Fluor 647 (actin, red), and anti-GFP Alexa Fluor 488 (PEDF, green). The slides were visualized using confocal microscopy at wavelengths 405 nm (DAPI), 647 nm (Phalloidin), and 488 nm (PEDF). The data presented are representative of three independent experiments with three samples/condition (n = 3). (b) Localization of VEGF receptors. HUVECs were plated at 1 × 105 cells/ml in serum free media and incubated overnight at 37 °C. Cells were then fixed and stained with primary and fluorescent-labeled secondary antibodies to detect localization of VEGFR-1 (A) and VEGFR-2 (D). Recombinant PEDF-6x-His-GFP fusion protein was added at 10 μg/ml alone (B, E) or with 25 ng/ml VEGF165 (C, F) and incubated at 37°C for 2 and 8 h (only 2 h shown for PEDF alone and 8 h shown for PEDF + VEGF). The cells were fixed at each time point and immunostained with goat anti-VEGFR-1 or VEGFR-2 followed by anti-goat Alexa Fluor 568 secondary. The nuclei were stained with DAPI (blue) and the actin was stained with Phalloidin Alexa Fluor 647. The slides were visualized using confocal microscopy at wavelengths 405 nm (DAPI), 647 nm (Phalloidin), and 568 nm (VEGFRs). The data presented are representative of one experiment with three samples/condition (n = 3).
Discussion
Anti-angiogenic activity of PEDF has been reported to arise from different types of activities, which may or may not be part of the same pathway. First, PEDF can block VEGF binding to receptors in vivo and in vitro (Cai et al. 2006; Ablonczy et al. 2009), leading to internalization and proteolysis of the receptors. Our results suggest that such a process may be the direct result of PEDF binding to the VEGF receptors, rather than interaction with VEGF itself. In our in vitro assay, recombinant PEDF directly bound strongly to VEGFR-1 and more weakly to VEGFR-2 (Fig. 3). Cai et al. found that VEGFR-1 engagement by PEDF may lead to secondary regulation of VEGFR-2 activity (Cai et al. 2006). Our determination of stronger binding affinity of PEDF for VEGFR-1 than VEGFR-2 provides support for this mechanism. In microscopic examination of treated cells, we do detect perinuclear accumulation, particularly of VEGFR-2 epitopes, after PEDF treatment. This appearance is suggestive of newly synthesized VEGFRs, as this perinuclear staining is also present in untreated cells, time 0 (Fig. 4b (A)) in addition to the dispersed receptors. VEGFRs are less apparent as dispersed signals in PEDF-treated cells, with or without VEGF treatment. This is consistent with findings by others that PEDF leads to intramembrane proteolysis and internalization of VEGF receptors after PEDF treatment (Cai et al. 2006, 2011). Interestingly, a recent report demonstrates that PEDF directly interacts with caveolin-1, which may suggest a mechanism for internalization of VEGFR-bound PEDF (Matsui et al. 2013). Secondly, expression of VEGF is downregulated by PEDF; the decrease in expression has been noted at the mRNA (Zhang et al. 2006) and protein (Zhang et al. 2011) levels. Zheng et al. (2010) showed that this is due to PEDF inhibition of JAK-STAT-mediated signaling. A recent study demonstrated transportin-mediated nuclear import of ectopically expressed PEDF, and suggested that anti-angiogenic activity of PEDF may require nuclear transport, either directly after synthesis or as a consequence of cellular uptake of secreted PEDF (Anguissola et al. 2011). Our results provide support for uptake of exogenous PEDF, as cells treated with both VEGF and PEDF showed some possible nuclear accumulation of PEDF 8 h after treatment (Fig. 4a (C, G)). This may have implications for the efficacy of therapeutic applications of exogenous PEDF.
Our studies suggest that extracellular PEDF is fully capable of mediating inhibitory effects on proliferation and migration of endothelial cells. Our recombinant PEDF-6×-HIS-GFP was functional in inhibition of VEGF-stimulated endothelial cell proliferation and migration through basement membrane (Fig. 2). The concentrations at which the recombinant PEDF affected these two processes were dramatically different, however. While migration of endothelial cells was inhibited at 1 ng/ml, proliferation was most completely inhibited at 100 μg/ml, although partial effects were noted at 100-fold lower concentrations (data not shown). This difference may be due to the engagement of different receptor species by PEDF, leading to different response profiles for the pathways affected by them. Interestingly, we noted an approximate 10-fold difference in binding of PEDF to VEGFR-1 versus VEGFR-2 in our in vitro binding assay (Fig. 3). Additionally, others have used 20-fold lower concentrations of PEDF to demonstrate inhibition of proliferation in the absence of VEGF (Subramanian et al. 2012), while our inclusion of VEGF may provide competition for PEDF activity. These investigators also demonstrated that there two distinct forms of PEDF; it is possible that the relative balance of these two forms may influence activity in different assays. The PEDF used by Duh et al. was also shown to be glycosylated (Duh et al. 2002), and our recombinant PEDF migrated at a higher than expected molecular weight (~90 kDa instead of the predicted 75 kDa) in SDS gels, suggesting the possibility of glycosylation of our PEDF as well. Difference in the extent and pattern of glycosylation could be responsible for these differences in activity. It is also important to consider that in the migration assay, VEGF is present in the bottom chamber as chemoattractant, while PEDF was added to cells in the upper chamber. This arrangement may have allowed PEDF to block the VEGF receptor prior to engagement of VEGF itself, thus blocking migration toward the attractant. VEGF was applied directly to treated cells in the viabilty/proliferation assay only 30 min after cells were treated with PEDF; this may have permitted more competition between PEDF and VEGF for receptor binding.
The morphological appearance of PEDF-treated HUVEC cells seems different from that of untreated cells. Although general cell size and shape were similar, untreated cells exhibited prominent networks of actin stress fibers (Fig. 4b (A, D)), while treated cells were less extended, and the stress fibers were more linearly aligned (Fig. 4a (A–C, E–G)). The more extended appearance was restored after 24 h post-treatment. The known ability of PEDF to bind the non-integrin laminin receptor (Bernard et al. 2009) may be responsible for this apparent difference in attachment characteristics of treated versus untreated cells. Binding to hyaluronan (Becerra et al. 2008) could potentially also affect treated cells’ matrix attachment.
Together, this array of effects on cells by recombinant PEDF could be due to engagement of a variety of known PEDF receptors. However, our study shows that it is possible that direct interaction of PEDF with VEGF receptors may be sufficient to block angiogenic activity.
References
Ablonczy Z, Prakasam A, Fant J, Fauq A, Crosson C, Sambamurti K (2009) Pigment epithelium-derived factor maintains retinal pigment epithelium function by inhibiting vascular endothelial growth factor-R2 signaling through gamma-secretase. J Biol Chem 284:30177–30186
Anguissola S, McCormack WJ, Morrin MA, Higgins WJ, Fox DM, Worrall DM (2011) Pigment epithelium-derived factor (PEDF) interacts with transportin SR2, and active nuclear import is facilitated by a novel nuclear localization motif. PLoS One 6:e26234
Becerra SP, Notario V (2013) The effects of PEDF on cancer biology: mechanisms of action and therapeutic potential. Nat Rev Cancer 13:258–271
Becerra SP, Perez-Mediavilla LA, Weldon JE, Locatelli-Hoops S, Senanayake P, Notari L, Notario V, Hollyfield JG (2008) Pigment epithelium-derived factor binds to hyaluronan. Mapping of a hyaluronan binding site. J Biol Chem 283:33310–33320
Bernard A, Gao-Li J, Franco CA, Bouceba T, Huet A, Li Z (2009) Laminin receptor involvement in the anti-angiogenic activity of pigment epithelium-derived factor. J Biol Chem 284:10480–10490
Cai J, Chen Z, Ruan Q, Han S, Liu L, Qi X, Boye SL, Hauswirth WW, Grant MB, Boulton ME (2011) {gamma}-secretase and presenilin mediate cleavage and phosphorylation of vascular endothelial growth factor receptor-1. J Biol Chem 286:42514–23
Cai J, Jiang WG, Grant MB, Boulton M (2006) Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J Biol Chem 281:3604–3613
Chen L, Zhang SS, Barnstable CJ, Tombran-Tink J (2006) PEDF induces apoptosis in human endothelial cells by activating p38 MAP kinase dependent cleavage of multiple caspases. Biochem Biophys Res Commun 348:1288–1295
Chuderland D, Hasky N, Ben-Ami I, Kaplan-Kraicer R, Grossman H, Shalgi R (2013) A physiological approach for treating endometriosis by recombinant pigment epithelium-derived factor (PEDF). Hum Reprod 28:1626–1634
Craword SE, Fitchev P, Veliceasa D, Volpert OV (2013) The many facets of PEDF in drug discovery and disease: a diamond in the rough or split personality disorder? Expert Opin Drug Discovery 8:769–792
Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, Bouck NP (1999) Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285:245–248
Deshpande M, Notari L, Subramanian P, Notario V, Becerra SP (2012) Inhibition of tumor cell surface ATP synthesis by pigment epithelium-derived factor: implications for antitumor activity. Int J Oncol 41:219–227
Doggett DL, Rotenberg MO, Pignolo RJ, Phillips PD, Cristofalo VJ (1992) Differential gene expression between young and senescent, quiescent WI-38 cells. Mech Ageing Dev 65:239–255
Duffy KE, Lamb RJ, Mateo LRS, Jordan JL, Canziam G, Brigham-Burke M, Korteweg J, Cunningham M, Beck HS, Carton J et al (2007) Down modulation of human TLR3 function by a monoclonal antibody. Cell Immunol 248:103–114
Duh EJ, Yang HS, Suzuma I, Miyagi M, Youngman E, Mori K, Katai M, Yan L, Suzuma K, West K et al (2002) Pigment epithelium-derived factor suppresses ischemia-induced retinal neovascularization and VEGF-induced migration and growth. Invest Ophthalmol Vis Sci 43:821–829
Gimbrone MA Jr, Leapman SB, Cotran RS, Folkman J (1972) Tumor dormancy in vivo by prevention of neovascularization. J Exp Med 136:261–276
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364
Hirsch J, Johnson CL, Nelius T, Kennedy R, Riese W, Filleur S (2011) PEDF inhibits IL8 production in prostate cancer cells through PEDF receptor/phospholipase A2 and regulation of NFkappaB and PPARgamma. Cytokine 55:202–210
Ho TC, Chen SL, Shih SC, Chang SJ, Yang SL, Hsieh JW, Cheng HC, Chen LJ, Tsao YP (2011) Pigment epithelium-derived factor (PEDF) promotes tumor cell death by inducing macrophage membrane tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol Chem 286:35943–35954
Ho TC, Chen SL, Yang YC, Liao CL, Cheng HC, Tsao YP (2007) PEDF induces p53-mediated apoptosis through PPAR gamma signaling in human umbilical vein endothelial cells. Cardiovasc Res 76:213–223
Hong H, Zhou T, Fang S, Jia M, Xu Z, Dai Z, Li C, Li S, Li L, Zhang T et al (2014) Pigment epithelium-derived factor (PEDF) inhibits breast cancer metastasis by down-regulating fibronectin. Breast Cancer Res Treat 148:61–72
Kanda S, Mochizuki Y, Nakamura T, Miyata Y, Matsuyama T, Kanetake H (2005) Pigment epithelium-derived factor inhibits fibroblast-growth-factor-2-induced capillary morphogenesis of endothelial cells through Fyn. J Cell Sci 118:961–970
Matsui T, Higashimoto Y, Taira J, Yamagishi S (2013) Pigment epithelium-derived factor (PEDF) binds to caveolin-1 and inhibits the pro-inflammatory effects of caveolin-1 in endothelial cells. Biochem Biophys Res Commun 441:405–410
Notari L, Arakaki N, Mueller D, Meier S, Amaral J, Becerra SP (2010) Pigment epithelium-derived factor binds to cell-surface F(1)-ATP synthase. FEBS J 277:2192–2205
Notari L, Baladron V, Aroca-Aguilar JD, Balko N, Heredia R, Meyer C, Notario PM, Saravanamuthu S, Nueda ML, Sanchez-Sanchez F et al (2006) Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J Biol Chem 281:38022–38037
Notari L, Miller A, Martinez A, Amaral J, Ju M, Robinson G, Smith LE, Becerra SP (2005) Pigment epithelium-derived factor is a substrate for matrix metalloproteinase type 2 and type 9: implications for downregulation in hypoxia. Invest Ophthalmol Vis Sci 46:2736–2747
Orgaz JL, Benguria A, Sanchez-Martinez C, Ladhani O, Volpert OV, Jimenez B (2011) Changes in the gene expression profile of A375 human melanoma cells induced by overexpression of multifunctional pigment epithelium-derived factor. Melanoma Res 21:285–297
Park K, Lee K, Zhang B, Zhou T, He X, Gao G, Murray AR, Ma JX (2011) Identification of a novel inhibitor of the canonical Wnt pathway. Mol Cell Biol 31:3038–3051
Spranger J, Osterhoff M, Reimann M, Mohlig M, Ristow M, Francis MK, Cristofalo V, Hammes HP, Smith G, Boulton M et al (2001) Loss of the antiangiogenic pigment epithelium-derived factor in patients with angiogenic eye disease. Diabetes 50:2641–2645
Steele FR, Chader GJ, Johnson LV, Tombran-Tink J (1993) Pigment epithelium-derived factor: neurotrophic activity and identification as a member of the serine protease inhibitor gene family. Proc Natl Acad Sci U S A 90:1526–1530
Subramanian P, Deshpande M, Locatelli-Hoops S, Moghaddam-Taaheri S, Gutierrez D, Fitzgerald DP, Guerrier S, Rapp M, Notario V, Becerra SP (2012) Identification of pigment epithelium-derived factor protein forms with distinct activities on tumor cell lines. J Biomed Biotechnol 2012:425907
Subramanian P, Locatelli-Hoops S, Kenealey J, Desjardin J, Notari L, Becerra SP (2013) Pigment epithelium-derived factor (PEDF) prevents retinal cell death via pigment epithelium-derived factor-R (PEDF-R): identification of a functional ligand binding site. J Biol Chem 288:23928–42
Subramanian P, Notario PM, Becerra SP (2010) Pigment epithelium-derived factor receptor (PEDF-R): a plasma membrane-linked phospholipase with PEDF binding affinity. Adv Exp Med Biol 664:29–37
Volpert OV, Zaichuk T, Zhou W, Reiher F, Ferguson TA, Stuart PM, Amin M, Bouck NP (2002) Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med 8:349–357
Zhang SX, Wang JJ, Gao G, Parke K, Ma JX (2006) Pigment epithelium-derived factor downregulates vascular endothelial growth factor (VEGF) expression and inhibits VEGF-VEGF receptor 2 binding in diabetic retinopathy. J Mol Endocrinol 37:1–12
Zhang Y, Han J, Yang X, Shao C, Xu Z, Cheng R, Cai W, Ma J, Yang Z, Gao G (2011) Pigment epithelium-derived factor inhibits angiogenesis and growth of gastric carcinoma by down-regulation of VEGF. Oncol Rep 26:681–686
Zheng Z, Chen H, Zhao H, Liu K, Luo D, Chen Y, Yang X, Gu Q, Xu X (2010) Inhibition of JAK2/STAT3-mediated VEGF upregulation under high glucose conditions by PEDF through a mitochondrial ROS pathway in vitro. Invest Ophthalmol Vis Sci 51:64–71
Acknowledgments
EKJ and JEK dedicate this study to their friend and colleague, Mary Kay Francis, who lost her battle with leukemia while the study was in progress. Mary Kay’s pioneering work on EPC-1/PEDF led to this research, and the authors treasured her enthusiasm and inspirational leadership. She is truly missed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: T. Okamoto
Rights and permissions
About this article

Cite this article
Johnston, E.K., Francis, M.K. & Knepper, J.E. Recombinant pigment epithelium-derived factor PEDF binds vascular endothelial growth factor receptors 1 and 2. In Vitro Cell.Dev.Biol.-Animal 51, 730–738 (2015). https://doi.org/10.1007/s11626-015-9884-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-015-9884-0