
Abstract
Introduction
Improvement in imaging has resulted in frequent diagnosis of benign and premalignant pancreatic tumors. Pancreatic nerve sheath (PNS) tumors are one of the rarest pancreatic tumors. Literature on PNS is limited and their biology is poorly understood. Here, we report the largest series of PNS tumors to date and review the literature to evaluate the current data available on PNS tumors.
Methods
An institutional database was used to identify patients who underwent resection for PNS tumors. Clinicopathological characteristics and outcomes of these patients were reported. Furthermore, a review of literature was performed.
Results
From January 1994 through December 2016, seven patients underwent resection for PNS tumors. The median age was 57.7 years (IQR, 44.9–61.9) and the sex was approximately equally distributed (male = 4; 57.1%). Three (42.9%) patients were diagnosed incidentally and six (85.7%) were misdiagnosed as having other pancreatic tumors. The median tumor size was 2.1 (IQR 1.8–3.0) cm and six (85.7%) had no nodal disease. At a median follow-up of 15.5 (IQR 13.7–49.3) months, six patients were alive without evidence of disease and one patient was lost to follow-up. The literature review identified 49 studies reporting 54 patients with PNS tumors. Forty-six were misdiagnosed as having other pancreatic tumors. The median tumor size was 3.6 (range 1–20) cm, nodal disease was present in six patients (22.2%), and no patient had distant metastatic disease. At the time of last follow-up, all patients were free of disease.
Conclusion
This is the largest single institution series on PNS tumors reported to date. These tumors are rare and are often misdiagnosed, given their radiological characteristics. PNS tumors have a benign course of disease and surgical resection results in favorable long-term outcomes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recent improvements in imaging techniques have led to an increase in the diagnosis of premalignant and benign tumors of the pancreas. One of the rarest reported type of pancreatic tumors is pancreatic nerve sheath (PNS) tumor, more commonly known as pancreatic schwannomas (PS).
Schwanommas are tumors that arise from the Schwann cells surrounding nerves and are commonly found in the periphery of the upper and lower extremities, head and neck, peritoneum, and retroperitoneum.1 Schwann cells surrounding the vagus nerve that courses through the pancreas can lead to the formation of pancreatic schwannomas or neurofibromas.2 These, in addition to paragangliomas arising in the pancreas are collectively referred to as PNS tumors. These tumors were first described by Moller-Pedderson et al. in 1982, and to date, 49 articles have reported 54 cases in the literature; the largest series of three patients being reported by Ferrozi et al.1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49 These tumors are predominantly solid but can present with a variety of degenerative changes, including cystic features, calcification, hemorrhage, hyalinization, and xanthomatous infiltration.1 Given their rarity and the wide range of their radiological appearance, these tumors are often misdiagnosed. Furthermore, due to the lack of literature, the biology of PNS tumors is poorly understood.17 In this study, we report the largest series reported to date on PNS tumors and present the current literature available on this disease.
Methods
Study Design and Data Collection
A prospectively maintained, institutionally approved database on all patients undergoing pancreatic resection at the Johns Hopkins Hospital was reviewed to identify patients who underwent surgical resection for PNS tumors between 1994 and 2016. Clinicopathological data were extracted from the database, and missing data were collected from the patients’ electronic medical records. Continuous variables were reported as means and standard deviations or medians and ranges as deemed appropriate, while all categorical variables were reported as frequencies and percentages. All analyses were performed using STATA version 14.1 (StataCorp, College Station, TX).
Review of Literature
A review of literature through July 2016 was performed to identify studies reporting patients with PNS tumors. A comprehensive search was performed using PubMed, EMBASE, and Medline using the terms “Pancreatic nerve sheath tumors,” “Pancreatic schwannoma,” “Pancreatic paraganglioma,” “pancreatic neurinomas,” and “pancreatic neurilemomas.” Two authors (AAJ and KC) performed independent reviews and then compiled a comprehensive list of available literature. The references in the studies identified during the initial review were screened to identify more articles reporting patients with PNS tumors. In cases where multiple pancreatic tumors were reported, data relevant to patients with PNS were extracted. When applicable, data that reappeared in the review were cross verified across multiple articles for data accuracy. Studies in languages other than English were excluded. Studies were limited to publication through July 2016 to allow for adequate reporting. Data on the clinicopathological features (including age, gender, symptoms, tumor location, initial diagnosis, surgery type, tumor size, nodal status, and distant metastasis) and outcomes (postoperative complications, recurrence of disease, and overall survival) were collected. A complete list of variables can be seen in Tables 1, 2, 3 and 4.
The study was approved by the Institutional Review Board for Human Research and complied with all Health Insurance Portability and Accountability Act regulations.
Results
During the study period, seven patients underwent surgical resection for primary PNS tumors (Table 1). The median age was 57.7 years (IQR, 44.9–61.9) and sex was approximately equally distributed (N = 4 males, 57.1% vs. N = 3 females, 42.9%), while a majority were white (N = 6, 85.7%). The most common presenting symptoms included abdominal pain (N = 4, 57.1%) and jaundice (N = 2, 28.6%). Two (28.6%) patients were diagnosed incidentally on routine medical evaluation or imaging done for other diseases. A majority (N = 5, 71.4%) had no history of smoking. The patients’ past medical history was significant for hypertension (N = 3, 42.9%), hyperlipidemia (N = 2, 28.6%), and pancreatitis (N = 1, 14.3%). One (14.3%) patient had a history of prior bladder cancer and one patient (14.3%) had a family history significant for colorectal cancer. The patient with bladder cancer underwent surgical resection prior to the diagnosis of the PNS tumor.
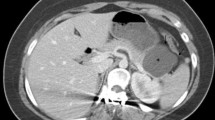
All patients were initially evaluated using a pancreas protocol computed tomography. (Table 2). Imaging demonstrated cystic features in six patients (85.7%) (Fig. 1). Specifically, nodularity was observed in two (28.6%) patients (Fig. 2), hypodensity was observed in two (28.6%) patients, central necrosis was observed in two (28.6%) patients (Fig. 3), while three (42.9%) patients had rim enhancement. The presumed diagnosis based on cross-sectional imaging included pancreatic cystic neoplasm (N = 3, 42.9%), intraductal papillary mucinous neoplasm (IPMN) (N = 1, 14.3%), pancreatic neuroendocrine tumor (N = 1, 14.3%), carcinoid tumor (N = 1, 14.3%), and lymphoma (N = 1, 11.1%). Subsequently five patients (71.4%) underwent evaluation via endoscopic ultrasound (EUS) and fine needle aspiration. The suggested diagnosed based upon histopathological review of the cytology included malignant neoplasm (N = 1, 14.3%), carcinoid tumor (N = 1, 14.3%), and PNS tumor (N = 1, 14.3%), and two (28.6%) patient’s biology was deemed to be inconclusive.

CT scan of a 62-year-old male demonstrating a 3.8-cm mass arising off the medial aspect of the uncinate process. No intrahepatic or pancreatic duct dilatation was observed. The mass was well defined but had cystic features. Furthermore, there was peripheral enchantment of the lesion. Presumed diagnosis was a pancreatic cystic lesion

CT scan of a 57-year-old female demonstrating a 1.9-cm hypodense, cystic mass arising from the neck of the pancreas. This lesion was slightly denser than simple cyst, and had mural nodularity. Presumed diagnosis was a pancreatic cystic neoplasm

CT scan of a 61-year old female demonstrating a 8.4-cm well-circumscribed, homogenous mass arising from the head of the pancreas, with upstream pancreatic duct dilatation. The mass had several smaller foci of internal cystic changes with central necrosis. Presumed diagnosis was a pancreatic cystic neuroendocrine tumor
A majority (N = 4, 57.1%) of the tumors were located in the head of the pancreas. Six patients (85.7%) underwent pancreaticoduodenectomy while one patient (14.3%) underwent a central pancreatectomy. The final histopathological examination confirmed a diagnosis of PNS tumors in seven patients (100%) (Table 3). The median size of the tumor was 2.1 cm (IQR 1.6–3.5), and a majority had no nodal disease (N = 6, 85.7%). The one-node-positive patient (14.3%) was found to have metastatic granulocytic paraganglioma in one lymph node. A negative margin was achieved in all seven (100%) patients. Three patients (42.9%) each had a T1 and T2 tumor while one patient (14.3%) had a T3 tumor. No patients were found to have metastatic disease. The patients were classified as having AJCC stage IA (N = 1, 14.3%), IB (N = 3, 42.9%), IIA (N = 1, 14.3%), and IIB (N = 2, 28.6%) disease as defined by the AJCC 7th edition.50
The median length of hospitalization was 7 days (IQR 7–10), and three patients (42.9%) developed postoperative complications. One patient (14.3%) experienced delayed gastric emptying, while one patient (14.3%) developed a postoperative pancreatic fistulae (POPF) (Table 3).
The median follow-up was 15.5 months (IQR 13.7–49.3). At the time of most recent follow-up, six patients (85.7%) were alive without evidence of disease and one patient (14.3%) was lost to follow-up.
Review of Literature
The review of literature yielded 1526 potential articles. After excluding those reporting PNS of origin other than the pancreas, and those with overlapping patients, a total of 49 studies reporting 54 patients with PNS tumors were identified. (Table 4) The patient population was approximately equally distributed with regard to sex (31 females, 57.4% vs. 23 males, 42.6%) and the median age was 55 years (range 20–87). The presenting symptoms included abdominal pain (N = 28, 51.9%), weight loss (N = 10, 18.5%), dyspepsia (N = 4, 7.4%), and jaundice (N = 1, 1.8%), while 24 patients (44.4%) were diagnosed incidentally. The tumor was more commonly located in the head of the pancreas (N = 23, 42.6%), while 15 patients (27.8%) had tumors in the body of the pancreas.
The presumed diagnosis was reported for 46 patients (85.2%), of whom only three (5.6%) had a presumed diagnosis of PNS. For the remaining patients, it varied from pancreatic cystic neoplasm (N = 19, 35.2%) to pancreatic ductal adenocarcinoma (N = 3, 5.6%), MCN (N = 6, 11.2%), microcystic adenoma (N = 1, 1.9%), pancreatic neuroendocrine tumor (PNET) (N = 8, 14.8%), and pseudocyst (N = 4, 7.4%).
Type of surgery was reported for 48 patients (88.9%) of whom 19 (35.2%) underwent pancreaticoduodenectomy, 11 (20.4%) underwent distal pancreatectomies, nine (16.7%) received enucleations, and one (1.9%) received a central pancreatectomy. Seven patients (13.0%) underwent nonspecific pancreatectomies, while one was deemed to have unresectable disease and underwent a palliative gastrojejunostomy.8
On histopathological review, the median size of the tumor (reported on 54 patients) was 3.6 cm (range 1–20). Nodal disease was present in six patients (22.2%) (reported on 27 patients), while no patients had distant metastatic disease. At a median follow-up of 15.5 (range 6–24) months, no patients had recurrence of disease.
Discussion
PNS tumors represent an exceedingly rare tumor of the pancreas.2They are generally benign neoplasms of pancreatic nerve sheaths originating on the vagus nerve and are often misdiagnosed as cystic pancreatic tumors.2 On gross histopathological examination, these tumors are generally well-differentiated, tan-yellow colored, round, and encapsulated, and can undergo degenerative changes such as hyalinization, cystic degeneration, hemorrhage, and xanthomatous infiltration. These tumors stain strongly for the S100 protein, vimentin, and CD56 + .12,51 Additionally, they stain for CD34-, CD117-, DOG1-, and AE1-/AE3-.12 Commonly, PNS tumors are negative for cytokeratin, c-kit, desmin, alpha smooth muscle actin, and smooth muscle myosin.1,30 Furthermore, cytogenetic studies have shown that most PNS tumors generally have mutations resulting in monosomy 22 or loss of 22q.1,20
To date, only 54 cases have been documented in English literature with the largest series reporting three cases.2 Together with our present series, which is the largest to date, a total of 63 cases of PNS tumors have been identified. The combined cohort demonstrates that PNS tumors show no preference for age or gender. Patients are typically asymptomatic or present with generalized symptoms including abdominal pain and weight loss. The median tumor size for all 61 cases was 2.9 cm and a majority (N = 27, 44.3%) were located in the head of the pancreas. Nodal disease was present in 4 (6.6%) of these patients. This is a clinically relevant finding given that while it is thought that all PNS tumors are benign, in fact, some of them may have malignant potential as evident by the nodal involvement, which warrants surgical resection rather than observation. Furthermore, if patients are symptomatic from their tumor, or the tumor growth is observed on subsequent scans, surgical resection is recommended.
On cross-sectional imaging, these tumors often mimic pancreatic cystic lesions with calcifications.1 Typically, imaging demonstrates encapsulated and well-defined lesions while endoscopic ultrasound (EUS) demonstrates hypoechoic masses. Unfortunately, fine-needle aspiration (FNA) is unreliable for PNS tumors, in part, due to the hypocellularity.42 Only a small minority (N = 4, 6.6%) of cases were diagnosed as PNS tumors preoperatively using EUS-guided FNA.8,15,25 Diagnostic challenges also stem from the heterogeneity of samples, as PNS tumors may contain epithelioid, inflammatory, or reactive cells.42 Microscopically, PNS contain two areas: Antoni A areas and Antoni B areas. Antoni A areas are characterized by hypercellular regions of tightly packed, monomorphic, spindle-shaped Schwann cells. The cells in Antoni A areas notably display nuclear palisading.51 Antoni B areas are characterized by hypocellular regions where the aforementioned degenerative changes are generally observed.51 Further, the Schwann cells in Antoni B areas tend to appear with nuclei that are suspended in a myxoid, microcystic matrix. There are multiple cases in the literature that have shown varying proportions of Antoni A and B areas in PNS tumors.51 Tumors with a high Antoni A/Antoni B ratio appear non-homogenous due to increased lipid content, while tumors with a low ratio appear hypodense, cystic, and multiseptated. With contrast imaging, Antoni A areas will show enhancement due to increased vascular involvement, while Antoni B areas will be non-enhancing due to less vascularity. Importantly, the diagnosis of PNS tumor does not preclude other cancer diagnoses, as concurrent PNS tumor and PDAC have been observed.
In both our institutional experience and literature, all cases of resected PNS tumors achieved good long-term outcomes; none of the patients developing recurrence at the last follow-up. PNS tumors are generally benign, and are believed to be relatively indolent tumors. A diagnosis of PNS tumor should be considered in the setting of heterogeneous masses of the pancreas particularly with calcification, hemorrhage, or hyalinization. Accurate diagnosis of pancreatic cystic lesions remains inexact; in fact, up to 25% of IPMNs are misdiagnosed preoperatively.52 As such, improved diagnostic imaging remains a need for accurate diagnosis of pancreatic tumors. Future studies may seek to rely on mutational analysis of preoperative FNA biopsies of these tumors. This study has several limitations, as is common with all retrospective analyses, including the potential for inaccurate data reporting and missed studies for inclusion.
In conclusion, PNS tumors are exceptionally rare tumors of the pancreas that are often misdiagnosed as other cystic pancreatic tumors. Surgical resection remains the treatment of choice and can help achieve excellent long-term outcomes.
References
Duma, N., et al., Enlarging Pancreatic Schwannoma: A Case Report and Review of the Literature. Clin Pract, 2015. 5(4): p. 793.
Ferrozzi, F., D. Bova, and G. Garlaschi, Pancreatic schwannoma: report of three cases. Clin Radiol, 1995. 50(7): p. 492–5.
Abu-Zaid, A., et al., Pancreatic tail schwannoma in a 44-year-old male: a case report and literature review. Case Rep Oncol Med, 2013. 2013: p. 416713.
Aggarwal, G., et al., Rare asymptomatic presentations of schwannomas in early adolescence: three cases with review of literature. Int J Surg, 2010. 8(3): p. 203–6.
Akiyoshi, T., et al., Melanotic schwannoma of the pancreas: report of a case. Surg Today, 2004. 34(6): p. 550–3.
Almo, K.M. and L.W. Traverso, Pancreatic schwannoma: an uncommon but important entity. J Gastrointest Surg, 2001. 5(4): p. 359–63.
Brown, S.Z., et al., Schwannoma of the pancreas: a report of two cases and a review of the literature. Mod Pathol, 1998. 11(12): p. 1178–82.
Bui, T.D., et al., Pancreatic schwannoma. A case report and review of the literature. JOP, 2004. 5(6): p. 520–6.
Ciledag, N., K. Arda, and M. Aksoy, Pancreatic schwannoma: A case report and review of the literature. Oncol Lett, 2014. 8(6): p. 2741–2743.
Coombs, R.J., Case of the season. Malignant neurogenic tumor of duodenum and pancreas. Semin Roentgenol, 1990. 25(2): p. 127–9.
David, S. and J.S. Barkin, Pancreatic schwannoma. Pancreas, 1993. 8(2): p. 274–6.
Di Benedetto, F., et al., Pancreatic schwannoma of the body involving the splenic vein: case report and review of the literature. Eur J Surg Oncol, 2007. 33(7): p. 926–8.
Dorsey, F., M.W. Taggart, and W.E. Fisher, Image of the month. Pancreatic schwannoma. Arch Surg, 2010. 145(9): p. 913–4.
Eggermont, A., et al., Solitary malignant schwannoma of the pancreas: report of a case and ultrastructural examination. J Surg Oncol, 1987. 36(1): p. 21–5.
Fasanella, K.E., K.K. Lee, and N. Kaushik, Clinical challenges and images in GI.Benign schwannoma of the pancreatic head. Gastroenterology, 2007. 132(2): p. 489, 830.
Feldman, L., et al., Pancreatic schwannoma: report of two cases and review of the literature. Pancreas, 1997. 15(1): p. 99–105.
Gupta, A., et al., Pancreatic schwannoma: literature review. J Surg Educ, 2009. 66(3): p. 168–73.
Hirabayashi, K., et al., Cytological features of the cystic fluid of pancreatic schwannoma with cystic degeneration. A case report. JOP, 2008. 9(2): p. 203–8.
Hsiao, W.C., P.W. Lin, and K.C. Chang, Benign retroperitoneal schwannoma mimicking a pancreatic cystic tumor: case report and literature review. Hepatogastroenterology, 1998. 45(24): p. 2418–20.
J, D., et al., Pancreatic schwannoma - a rare case report. J Clin Diagn Res, 2014. 8(7): p. FD15–6.
Kameyama, N., et al., Single-port transumbilical laparoscopic excision of retroperitoneal schwannoma mimicking a nonfunctional endocrine tumor in the body of the pancreas: a case report. Surg Innov, 2013. 20(6): p. NP30–4.
Kim, G., et al., Pancreatic benign schwannoma: combined with hemorrhage in an internal cyst. J Dig Dis, 2011. 12(2): p. 138–41.
Kinhal, V.A., et al., Pancreatic schwannoma: Report of a case and review of literature. Indian J Surg, 2010. 72(Suppl 1): p. 296–8.
Lee, J.S., et al., Ancient schwannoma of the pancreas mimicking a cystic tumor. Virchows Arch, 2001. 439(5): p. 697–9.
Li, S., et al., Intrapancreatic schwannoma diagnosed by endoscopic ultrasound-guided fine-needle aspiration cytology. Diagn Cytopathol, 2009. 37(2): p. 132–5.
Liegl, B., et al., Microcystic/reticular schwannoma of the pancreas: a potential diagnostic pitfall. Pathol Int, 2011. 61(2): p. 88–92.
Liessi, G., et al., CT and MR imaging of melanocytic schwannomas; report of three cases. Eur J Radiol, 1990. 11(2): p. 138–42.
Melato, M., et al., The schwannoma: an uncommon type of cystic lesion of the pancreas. Ital J Gastroenterol, 1993. 25(7): p. 385–7.
Moller Pedersen, V., A. Hede, and N. Graem, A solitary malignant schwannoma mimicking a pancreatic pseudocyst. A case report. Acta Chir Scand, 1982. 148(8): p. 697–8.
Moriya, T., et al., Pancreatic schwannoma: Case report and an updated 30-year review of the literature yielding 47 cases. World J Gastroenterol, 2012. 18(13): p. 1538–44.
Mourra, N., J. Calvo, and L. Arrive, Incidental Finding of Cystic Pancreatic Schwannoma Mimicking a Neuroendocrine Tumor. Appl Immunohistochem Mol Morphol, 2016. 24(2): p. 149–50.
Mummadi, R.R., et al., Pancreatic Schwannoma presenting as a cystic lesion. Gastrointest Endosc, 2009. 69(2): p. 341; discussio 341.
Novellas, S., et al., MRI features of a pancreatic schwannoma. Clin Imaging, 2005. 29(6): p. 434–6.
Ohbatake, Y., et al., A case of pancreatic schwannoma - The features in imaging studies compared with its pathological findings: Report of a case. Clin J Gastroenterol, 2014. 7(3): p. 265–70.
Okuma, T., et al., Pancreatic schwannoma: report of a case. Surg Today, 2008. 38(3): p. 266–70.
Oshima, M., S. Yachida, and Y. Suzuki, Pancreatic schwannoma in a 32-year-old woman mimicking a solid-pseudopapillary neoplasm. Clin Gastroenterol Hepatol, 2010. 8(1): p. e1–2.
Paranjape, C., et al., Clinical characteristics, treatment, and outcome of pancreatic Schwannomas. J Gastrointest Surg, 2004. 8(6): p. 706–12.
Poosawang, W. and P. Kiatkungwankai, Pancreatic schwannoma: A case report and review of literature. J Med Assoc Thai, 2013. 96(1): p. 112–6.
Soumaoro, L.T., et al., Benign schwannoma of the pancreas. J Gastrointest Surg, 2005. 9(2): p. 288–90.
Stojanovic, M.P., et al., Malignant schwannoma of the pancreas involving transversal colon treated with en-bloc resection. World J Gastroenterol, 2010. 16(1): p. 119–22.
Suzuki, S., et al., Pancreatic schwannoma: a case report and literature review with special reference to imaging features. JOP, 2010. 11(1): p. 31–5.
Tafe, L.J. and A.A. Suriawinata, Cystic pancreatic schwannoma in a 46-year-old man. Ann Diagn Pathol, 2008. 12(4): p. 296–300.
Tan, G., et al., Cystic schwannoma of the pancreas. Ann Diagn Pathol, 2003. 7(5): p. 285–91.
Tofigh, A.M., et al., Rare presentation of pancreatic schwannoma: a case report. J Med Case Rep, 2008. 2: p. 268.
Urban, B.A., et al., CT findings in cystic schwannoma of the pancreas. J Comput Assist Tomogr, 1992. 16(3): p. 492–3.
von Dobschuetz, E., et al., Giant ancient schwannoma of pancreatic head treated by extended pancreatoduodenectomy. Pancreatology, 2004. 4(6): p. 505–8.
Walsh, M.M. and K. Brandspigel, Gastrointestinal bleeding due to pancreatic schwannoma complicating von Recklinghausen’s disease. Gastroenterology, 1989. 97(6): p. 1550–1.
Wu, W., et al., A Contemporary Evaluation of the Cause of Death and Long-Term Quality of Life After Total Pancreatectomy. World J Surg, 2016. 40(10): p. 2513–8.
Yu, R.S. and J.Z. Sun, Pancreatic schwannoma: CT findings. Abdom Imaging, 2006. 31(1): p. 103–5.
Edge, S.B. and C.C. Compton, The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol, 2010. 17(6): p. 1471–4.
Weiss, S.W., J.M. Langloss, and F.M. Enzinger, Value of S-100 protein in the diagnosis of soft tissue tumors with particular reference to benign and malignant Schwann cell tumors. Lab Invest, 1983. 49(3): p. 299–308.
Barron, M.R., et al., Does preoperative cross-sectional imaging accurately predict main duct involvement in intraductal papillary mucinous neoplasm? J Gastrointest Surg, 2014. 18(3): p. 447–55; discussion 5455-6.
Author information
Authors and Affiliations
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Javed, A.A., Wright, M.J., Hasanain, A. et al. Pancreatic Nerve Sheath Tumors: a Single Institutional Series and Systematic Review of the Literature. J Gastrointest Surg 24, 841–848 (2020). https://doi.org/10.1007/s11605-019-04201-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-019-04201-4