
Abstract
Hepatocellular carcinoma has been described to exhibit characteristics similar to that of neuroendocrine tumors (NETs). This includes similar anti-neoplastic responses to extracellular signal-regulated kinase (ERK) activation. NET cells and HepG2 cells have both shown growth inhibition with ERK activation. ZM336372, a Raf-1 activating agent, has been shown to cause growth inhibition and suppression of hormone secretion in a neuroendocrine cell line. Here we examine treatment of the HepG2 cell line with ZM336732 to determine if a similar anti-proliferative response will be obtained. HepG2 cells were treated with ZM336372 or solvent (dimethyl sulfoxide). The resulting effect on the proliferation was measured using the 3,4-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Western blot analysis was performed to examine the activation of the Raf-1/mitogen-activated protein kinase kinase/ERK pathway, chromogranin A production, and p21CIP1 level. Growth inhibition was observed with ZM336372 in a dose-dependent fashion. Minimal baseline phosphorylation of ERK 1/2 was observed; however, activation was observed after treatment with ZM336372. Chromogranin A secretion was suppressed due to treatment with ZM336372. A dose-dependent up-regulation of p21CIP1 was observed in response to ZM336372 treatment. ZM336372 causes growth inhibition, suppression of hormone secretion, and up-regulation of cell cycle inhibitors in a human hepatocellular carcinoma cell line, similar to that previously seen in NETs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer and a significant cause of cancer-related death throughout the world.1,2 The only effective forms of treatment for this disease are locoregional; systemic therapies are generally ineffective. Therefore, studies looking at cellular mechanisms of tumor growth are essential in order to discover efficacious therapies.
Examinations into the biology of HCC have shown characteristics similar to those of neuroendocrine tumors (NETs). HCC and NETs share a similar response to Notch-1 activation. In both NETs and HCC, Notch-1 activation causes growth inhibition and cell cycle arrest.3–5 This is unlike pancreatic adenocarcinoma, colon cancer, renal cell carcinoma, non-small cell lung cancer, and cervical cancer, where Notch1 activation causes cellular proliferation.6 In addition, chromogranin A secretion occurs in both NETs and HCC. Chromogranin A has long been used as a marker for NETs, but it has also been found to be elevated in the serum of patients with HCC.7
NETs and HCC uncommonly possess activating mutations of the Ras/Raf-1/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway 8–12 (Fig. 1), whereas activating mutations of this pathway are quite common in most other tumors.13,14 ERK pathway activation has been shown to cause growth inhibition in NET and HCC cell lines. Studies have shown that inducing Raf-1 expression in neuroendocrine cell lines leads to suppression of hormone secretion and cellular growth.15,16 Numerous other studies have also shown anti-neoplastic effects of activation of the Raf-1/MEK/ERK pathway on neuroendocrine tumors.17–19

Schematic representation of the Ras/Raf-1/MEK/ERK pathway.
Activation of ERK has previously been shown to cause growth inhibition in HepG2 cells due to treatment with hepatocyte growth factor (HGF) and induced constitutive expression of Ha-Ras.20 In addition, HGF treatment has been shown to result in up-regulation of the cyclin-dependent kinase inhibitors p21CIP1 and p27KIP1 leading to cell cycle arrest in response to ERK activation.21
ZM336372 is a small molecule discovered by screening a chemical library for a Raf-1 inhibitor. It was shown to inhibit Raf-1 in solution; however, when tested in cell culture, ZM336372 was shown to cause Raf-1 activation by greater than 100-fold.22 ZM336372 was previously tested in a neuroendocrine cell line. The H727 carcinoid tumor cell line was treated with ZM336372, causing diminished production of chromogramin A, suppression of cellular proliferation, and p21CIP1 up-regulation.23 Since ZM336372 has shown these effects in NETs and since NETs share a similar response to ERK activation as HCC, we decided to determine if Raf-1 activation due to ZM336372 treatment of a hepatocellular carcinoma cell line would result in anti-neoplastic effects similar to those previously seen in NETs.
Methods
Cell Culture
HepG2 cells (American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, VT, USA), 100 IU/ml of penicillin, and 100 pg/ml of streptomycin (Gibco). The cell lines were incubated in a humidified atmosphere of 5% CO2 at 37°C.
ZM336372 treatment
Treatment of the cell lines with ZM336372 (Tocris, Ellisville, MO, USA) was performed by addition of the appropriate concentration of drug from a 100 mM stock dissolved in 100% dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) to the culture media prior to adding the solution to cells plated the day before. The media and drug were exchanged every 2 days for the duration of the experiments.
MTT Assay
MTT reagent, 3,4-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma), was reconstituted in phosphate-buffered saline (PBS) to a final concentration of 5 mg/ml. MTT reagent was added to each culture being assayed in phenol red free media to equal one tenth the original culture volume and incubated for 3 h at 37°C. The medium was then removed, and the dye was solubilized with 350 μl of 0.1 N HCl in isopropanol. The absorbance was then measured at a wavelength of 540 nm with background subtraction at 630–690 nm.
Cellular Extracts
Media was aspirated, and the attached cells were washed once with PBS. PBS containing 0.5 M ethylenediamine tetraacetic acid was added to the cells, and then cells were removed using a cell scraper. Next, the cell suspension was centrifuged at 2,000 rpm for 5 min at 4°C. The supernatant was aspirated, and lysing buffer, consisting of Tris–sodium dodecyl sulfate buffer, phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail, was added to the cell pellets and incubated for 20–30 min at 4°C. After centrifugation at 1,300 rpm for 30 min at 4°C, supernatant was collected, and protein concentrations were performed using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA) following manufacturer’s instructions.
Western Blot Analysis
Approximately 40 μg of cellular extract from ZM336372-treated cells and controls were loaded onto pre-cast 10% polyacrylamide gels (Pierce). The gels were run at 100 V for 60 min and then transferred to polyvinylidene difluoride Immobilon-P membranes (Millipore, Bedford, MA, USA) at 40 V for 90 min. The membranes were blocked with a 5% milk solution for 1 h, and the primary antibodies, p-MEK 1/2, p-MAPK p44/p42, p-c-Raf, p21, and Cg A (Cell Signaling Technology, Beverly, MA, USA), were incubated overnight in bovine serum albumin (Sigma) at a 1:1,000 ratio. Following incubation with the primary antibody, membranes were washed three times for 5 min in Tris-buffered saline Tween-20 (TBS-T) wash buffer (Tris-buffered saline, 0.05% Tween 20). Next, goat anti-rabbit horseradish peroxidase-labeled antibody (Pierce) was added at ratio of 1:7,000 in milk solution and incubated for 1–2 h. The membranes were then washed again three times, for 5 min with TBS-T. SuperSignal West Pico Chemiluminescent Substrate (Pierce) was added according to manufacturer’s instructions and then incubated for 5 min. Following removal of the substrate, the membranes were placed in plastic sleeves and exposed to film. Anti-G3PDH antibody (Pierce) was utilized as a loading control at a ratio of 1:7,500.
MEK 1/2 inhibitor assay
Cell cultures were plated out at 1 million cells per 100 mm culture dish. Cells were pretreated with UO126 (Cell Signaling Technology), a MEK 1/2 inhibitor, for 45 min at a concentration of 5 μM. Following UO126 treatment, ZM336372 was added from a 100-mM stock solution in DMSO directly to the culture media in the culture dish at a concentration of 15 μM and mixed. Controls containing only UO126 and ZM336372 were also incubated with the experiment. Cellular extracts and Western blot were then performed as above.
Results
ZM336372 Causes Activation of Raf-1/MEK/ERK Cascade in HCC Cell Line
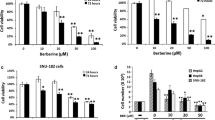
To clarify the cellular effects of ZM336372 on the Raf-1/MEK/ERK pathway in the HepG2 cell line, Western blot using antibodies targeted for the activated forms of these kinases was performed. Minimal phosphorylation of ERK 1/2 was observed at baseline (Fig. 2a). However, an increase in phosphorylated ERK 1/2 was observed after ZM336372 treatment in a dose-dependent fashion for the concentrations from 5 to 25μM. In addition, phosphorylation of Raf-1 and MEK 1/2 were also observed. This confirms that ZM336372 is able to cause activation of the Raf-1/MEK/ERK1/2 pathway in HepG2 cells.

Activation of the Raf-1/MEK/ERK pathway secondary to treatment with ZM336372. a Total cellular extracts from HepG2 cells treated with ZM336372 or control show a dose-dependent increase in ERK 1/2 activation following treatment with ZM336372 after 2 days. In addition, ZM336372 activation of Raf-1 and MEK 1/2 is shown. b Following treatment with UO126, the ZM336372-induced activation of ERK 1/2 is inhibited.
To determine if the ERK 1/2 activation by ZM336372 is dependent on phosphorylation of MEK 1/2, an inhibitor assay was performed using UO126. UO126 has previously been shown to prevent the phosphorylation of ERK 1/2 by both MEK1 and MEK 2. Since Raf-1 activation of ERK 1/2 is mediated through MEK 1/2, inhibition by UO126 should prevent ZM336372-mediated ERK phosphorylation if it occurs through this pathway. Incubation of HepG2 cells with UO126 prior to treatment with ZM336372 prevented the activation of ERK 1/2 by ZM336372 as demonstrated by Western blot in Fig. 2b. A concentration of 5 μM of UO126 was used to inhibit the ZM336372-induced phosphorylation of ERK 1/2. This correlates with the ZM336372-induced activation of ERK 1/2 being propagated through MEK 1/2.
ZM336372 Causes Growth Inhibition of the HepG2 Cell Line
The MTT assay was employed to determine if ZM336372 would cause an effect on cell growth, as previously described with HGF-induced ERK activation in this cell line.20 The MTT assay after 8 days of treatment (Fig. 3a) shows a decrease in growth rate in proportion to the concentration of ZM336372 added. Significant growth reduction is observed at the 5 μM concentration of ZM336372, with complete suppression of proliferation at the 15 μM concentration. This confirms that ZM336372 is able to cause suppression of proliferation in HCC cells.

Growth suppression secondary to treatment with ZM336372. a The MTT assay after 8 days of treatment shows a reduction in proliferation after treatment with the 5 μM concentration of ZM336372. Complete growth suppression of the HepG2 cells is observed with the 15 μM concentration. DMSO treated HepG2 cells were used as a control. b ZM336372 treatment of a pancreatic adenocarcinoma cell line results in no change in growth suppression. Panc-1 cells treated with 25μM of ZM336372 for 8 days showed no significant difference between cells treated with the DMSO control.
Since ZM336372 caused growth suppression in a cell line with minimal baseline ERK activation, the growth effects of ZM336372 when baseline ERK activation is present was investigated. Pancreatic adenocarcinomas commonly possess activating mutations of the Ras/Raf-1/MEK/ERK pathway and thus would not be expected to respond to ZM336372 treatment with growth suppression. The MTT assay was utilized to evaluate the growth effects of ZM336372 on Panc-1 cells. Figure 3b shows that no change in growth was noted between 25 μM of ZM336372 and control cells after 8 days of treatment. Above-baseline activation of MEK 1/2 and ERK 1/2 was seen in the pancreatic cancer cells on Western blot (data not shown).
ZM336372 Causes Up-regulation of the Cell Cycle Inhibitor p21CIP1
To determine if there is a ZM336372 treatment-induced effect on the cyclin-dependent kinase inhibitor p21CIP1, Western blot analysis was performed. Figure 4a shows a dose-dependent elevation in the expression of p21CIP1 in response to ZM336372 treatment. This correlates with previous studies showing a Raf-1-induced increase in p21CIP1 expression resulting in inhibition of proliferation.24 In addition, this also correlates with previous descriptions of ZM336372-induced cell cycle arrest in H727 cells and cell cycle arrest due to HGF treatment in HepG2 cells.

A. Up-regulation of p21CIP1 after ERK 1/2 activation by ZM336372. Minimal p21CIP1 is observed at baseline. Following 2 days of treatment with ZM336732, a dose-dependent increase in the level of p21CIP1 is observed. b Chromogranin A secretion is suppressed following ZM336372 treatment. A low level of Cg A is present at baseline in HepG2 cells. This is suppressed by treatment with ZM336372. Suppression of Cg A is observed at the 10μM dose.
ZM336372 Treatment Results in Suppression of Hormone Secretion
Chromogranin A secretion is a marker of neuroendocrine differentiation but has also been observed in the serum of patients with HCC. Previously, ZM336372 treatment in H727 cells resulted in decreased chromogranin A secretion. Figure 4b shows confirmation of chromogranin A secretion along with a dose-dependent decrease in hormone production in HepG2 cells, similar to that seen with carcinoid cells.
Discussion
The Raf-1/MEK/ERK pathway controls cellular differentiation, proliferation, and survival. Numerous tumor types contain activating mutations of this pathway resulting in neoplastic transformation. However, in other tumor types that typically do not harbor activating mutations of this pathway, growth inhibition has been described in response to activation of ERK 1/2.25
In a study using an estrogen-inducible Raf-1 construct in NIH 3T3 cells, Raf-1 activation was shown to induce p21CIP1 expression and elicit G1 arrest.24 In this same study, it is interesting to note that A-Raf activation led to cell cycle progression. Using a similar construct, activation of Raf-1 in NET cell lines has shown anti-neoplastic effects, including growth inhibition, decrease in hormone secretion, and morphologic changes.15,16 This sparked the interest in ZM336372 as a Raf-1 activating agent for treatment of NETs. Previously, treatment of the H727 NET cell line with ZM336372 has resulted in growth inhibition, a decrease in hormone secretion, and p21CIP1 up-regulation.23
In the HepG2 cell line, previous studies have shown a similar response to ERK 1/2 activation as compared to NETs. Ha-Ras induction leads to suppression of proliferation through ERK 1/2 in HepG2 cells.20 It is interesting to note that partial inhibition with the MEK inhibitor PD98059, after Ha-Ras induction, leads to growth proliferation. This suggests that the proliferation response may be a dose-related effect of ERK 1/2 activation. In addition, treatment of HepG2 cells with HGF also led to ERK 1/2 activation and resulted in inhibition of cellular proliferation.20 Activation of ERK 1/2 via HGF treatment resulted in up-regulation of p21CIP1 and p27KIP1 and cell cycle arrest at G1.21
Here we show that ZM336372 can also cause growth inhibition in HepG2 cells. Raf-1/MEK/ERK pathway activation resulted as expected from ZM336372 treatment. Inhibition of this pathway via UO126 proves that the ERK 1/2 activation of ZM336372 is through MEK 1/2. The induced expression of p21CIP1 seen here is similar to that seen previously with ZM336372 treatment and also similar to that seen with HGF, suggesting a possible mechanism for the observed suppression of proliferation.
In addition, CgA secretion was observed to be decreased in HepG2 cells in response to ZM336372 treatment. This is similar to that seen previously with the H727 cell line. In NET cells, CgA has been used as a marker for neuroendocrine differentiation. It has been shown that this suppression of Cg A has also been associated with hASH-1 down-regulation, but the exact mechanism by which this decrease in hormone secretion takes place has yet to be identified.
Conclusion
ZM336372 induces anti-neoplastic effects in HepG2 cells similar to those previously seen with ERK activation in this cell line and seen with NET cell lines. These data suggest that treatments that activate the ERK pathway may be effective in controlling this disease. Further studies with ZM336372 and other ERK activators are warranted as potential new agents in cancer therapy.
References
Blum HE. Hepatocellular carcinoma: therapy and prevention. World J Gastroenterol 2005;11:7391–7400.
Zhu AX. Hepatocellular carcinoma: Are we making progress? Cancer Invest 2003;21:418–428.
Nakakura EK, Sriuranpong VR, Kunnimalaiyaan M et al. Regulation of neuroendocrine differentiation in gastrointestinal carcinoid tumor cells by notch signaling. J Clin Endocrinol Metab 2005;90:4350–4356.
Qi R, An H, Yu Y et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res 2003;63:8323–8329.
Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J Biol Chem 2006;281:39819–39830.
Miyamoto Y, Maitra A, Ghosh B et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003;3:565–576.
Leone N, Pellicano R, Brunello F et al. Elevated serum chromogranin A in patients with hepatocellular carcinoma. Clin Exp Med 2002;2:119–123.
Younes N, Fulton N, Tanaka R et al. The presence of K-12 ras mutations in duodenal adenocarcinoma and the absence of absence of ras mutations in other small bowel adenocarcinomas and carcinoid tumors. Cancer 1997;79:1804–1808.
Yashiro T, Fulton N, Hara H et al. Comparison of mutations of ras oncogene in human pancreatic exocrine and endocrine tumors. Surgery 1993;114:758–763.
Parakevakou H, Saetta A, Skandalis K et al. Morphological-histochemical study of intestinal carcinoids and K-ras mutation analysis in appendiceal carcinoids. Pathol Oncol Res 1999;5:205–210.
Hoshino R, Chutani Y, Yamori T et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999;18:813–822.
Zeng JZ, Wang HY, Chen ZJ et al. Molecular cloning and characterization of a novel gene which is highly expressed in hepatocellular carcinoma. Oncogene 2002;21:4932–4943.
Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther 2005;4:677–685.
Polsky D, Cordon-Cardo C. Oncogenes in melanoma. Oncogene 2003;22:3087–3091.
Sippel RS, Chen H. Activation of the ras/raf-1 signal transduction pathway in carcinoid tumor cells results in morphologic transdifferentiation. Surgery 2002;132:1035–1039.
Sippel RS, Carpenter JE, Kunnimalaiyaan M et al. Raf-1 activation suppresses neuroendocrine marker and hormone levels in human gastrointestinal carcinoid cells. Am J Physiol Gastrointest Liver Physiol 2003;285:G245–254.
Nakagawa T, Mabry M, De Bustros A et al. Introduction of v-Ha-ras oncogene induces differentiation of cultured human medullary thyroid carcinoma cells. Proc Natl Acad Sci U S A 1987;84:5923–5927.
Ravi RK, Thiagalingam A, Weber E, McMahon M et al. Raf-1 causes growth suppression and alteration of neuroendocrine markers in DMS53 human small cell lung cancer cells. Am J Respir Cell Mol Biol 1999;20:543–549.
Chen H, Carson-Walter EB, Baylin SB, Nelkin BD et al. Differentiation of medullary thyroid cancer by c-Raf-1 silences expression of the neural transcription factor human acheate-scute homolog-1. Surgery 1996;120:168–172.
Tsukada Y, Miyazawa K, Kitamura N. High intensity ERK signal mediates hepatocyte growth factor-induced proliferation inhibition of the human hepatocellular carcinoma cell line HepG2. J Biol Chem 2001;276:40968–40976.
Han J, Tsukada Y, Hara E et al. Hepatocyte growth factor induces redistribution of p21CIP1 and p27KIP1 through ERK-dependent p16INK4a up-regulation, leading to cell lcycle arrest at G1 in HepG2 hepatoma cells. J Biol Chem 2005;280:31548–31556.
Hall-Jackson CA, Eyers PA, Cohen P et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem & Biol 1999;6:559–568.
Van Gompel JJ, Kunnimalaiyaan M, Holen K, Chen H. ZM336372, a Raf-1 activator, suppresses growth and neuroendocrine hormone levels in carcinoid tumor cells. Mol Cancer Ther 2005;4:910–917.
Woods D, Parry D, Cherwinski H et al. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21CIP1. Moll Cell Biol 1997;17:5598–5611.
Kunnimalaiyaan M, Chen H. The Raf-1 pathway: a molecular target for treatment of select neuroendocrine tumors. Anticancer Drugs 2006;17:139–142.
Acknowledgment
The authors would like to thank Lab of Herb Chen and the UW Analytical Lab. This work was supported in part by a Research Scholars Grant from the American Cancer Society, National Institutes of Health grants DK064735and CA109053; the George H.A. Clowes, Jr., Memorial Research Career Development Award of the American College of Surgeons, the Carcinoid Cancer Foundation Award (HC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Deming, D., Geiger, P., Chen, H. et al. ZM336372, A Raf-1 Activator, Causes Suppression of Proliferation in a Human Hepatocellular Carcinoma Cell Line. J Gastrointest Surg 12, 852–857 (2008). https://doi.org/10.1007/s11605-008-0495-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-008-0495-x