
Abstract
A stable and high oxygen permeable membrane system has been developed via partial replacement of iron with zirconium in SrCo0.8Fe0.2O3−δ compound. The initial powder series of composition SrCo0.8Fe0.2−yZryO3−δ (0 ≤ y ≤ 0.1), synthesized by an oxalate-based sol-gel route, is shown to exhibit a single perovskite-type cubic phase at 27 °C, transform to a brownmillerite (Ca2AlFeO5)-type orthorhombic structure, and finally assume a stable open cubic phase with disordered oxygen vacancies at temperatures determined by zirconium content, transitions being at 200 and 800 °C, respectively for the system y = 0.05. The highest JO2 of 2.54 × 10−6 mol cm−2 s−1 is realized in optimum composition of y = 0.05 at 1000 °C. The formation of cubic phase in Sr–Co–Fe–O system with abundance of disordered anion vacancies is crucial for both the high oxygen permeability and extended operational stability at elevated temperatures.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The development of membranes with high oxygen permeability and adequate stability is desirable in highly efficient oxygen separation systems [1]. The perovskite-type oxides with general formula ABO3 (A—rare earth or alkaline rare earth metal ions {Ba, Ca, Sr, La, etc.}; B—transition and other metal ions {Co, Fe, Zn, Mn, Ga, etc.}; O—oxygen ions) have been explored over the years for their oxygen permeation capacity and stability [2,3,4,5]. Their salient features include (i) a wide range of oxygen deficiency/vacancy, (ii) variable oxidation states of ions at A and/or B site, and (iii) mixed ionic and electronic conductivity. Consequently, ABO3−δ (A = Sr, La; B = Co, Fe; δ = oxygen deficiency) perovskites have been studied widely for a variety of applications, viz., gas/oxygen separation as membrane, oxygen pumps, solid oxide fuel cells, oxidative coupling of methane as catalyst, oxygen sensors, etc. [6,7,8,9]. Teraoka et al. [10, 11] synthesized Ln1−xAxCo1−yByO3−δ (Ln = La, Pr, Nd, Sm, Gd; A = Sr, Ca, Ba; B = Mn, Cr, Fe, Co, Ni, Cu) (x = 0–0.4, y = 0–0.2) by solid-state reaction of metal acetates and nitrates at elevated temperatures (970–1300 °C) and measured their oxygen permeability (JO2). Among them, SrCo0.8Fe0.2O3−δ membrane (thickness ~ 1 mm) showed the highest JO2 of 2.31 × 10−6 mol cm−2 s−1at 850 °C, but displayed limited-phase stability and mechanical steadiness [12,13,14,15,16]. Its noticeable high JO2 sustained interest in this ABO3–δ system with continual investigations [17,18,19,20,21]. Consequently, the partial replacement of B species by zirconium (Zr) in SrCo0.8Fe0.2O3−δ is reported to stabilize the perovskite-type cubic structure and induce disordering in oxygen vacancies [22, 23]. Also, the enhanced oxygen permeability in La0.2Ba0.8Co0.8Fe0.2−xZrxO3−δ (x = 0.05) at elevated temperatures is attributed to prevailing disorder in anion vacancies [24]. Zirconium in BaCo0.7Fe0.3−xZrxO3−δ (x = 0–0.12) is found to increase JO2 up to x = 0.06 (value being 2.7 ml min−1 cm−2 at 925 °C for 1-mm-thick membrane) but decrease at higher content [25]. Further, ZrO2 containing SrCo0.4Fe0.6O3–δ membrane displays high oxygen permeability (JO2) and remains stable as well at elevated temperatures and reduced oxygen partial pressures [26]. In contrast, Yang et al. [22, 23] found improved phase stability in SrCo0.4Fe0.6−xZrxO3−δ (x = 0–0.2) membranes, but with noticeable decrease in oxygen permeability. Chen et al. [27] and Lu et al. [28] also observed analogous results (i.e., improved structural stability and reduced oxygen permeability) in Zr-doped SrCo0.8Fe0.2O3−δ and Ba0.5Sr0.5Co0.8Fe0.2O3−δ membranes, produced with powders prepared by EDTA and solid-state reaction method, respectively. Obviously, zirconium incorporation in SrCo0.8Fe0.2O3−δ compound has led to conflicting oxygen permeability data which need to be resolved by further study. Thus, SrCo0.8Fe0.2−yZryO3−δ (y = 0–0.1) system becomes the natural choice and preferred for the present investigation. The objective has been to understand the effect of zirconium on its phase stability and oxygen permeability characteristics. The stability of perovskite structures is supposed to improve with addition of fixed valence state elements [27]. Also, increase in unit cell size (i.e., realizing a somewhat open structure) may facilitate movement of anions and enhance oxygen permeability. With fixed valency (4 +) and ionic radii larger than cobalt/iron species, besides being robust, zirconium is selected for incorporation in SrCo0.8Fe0.2O3−δ to yield desirable material for oxygen permeable membrane application.
Solid-state reaction, citrate-EDTA complexing method, and acetic acid-based sol-gel route have been used over the years for synthesis of oxide powders for membrane purposes [29,30,31,32]. Among them, sol-gel is optimal and offers certain advantages, viz., homogeneous mixing of precursors at atomic level, better stoichiometry control, high purity, and low cost [32]. Hence, an oxalate-based sol-gel route is chosen here to prepare SrCo0.8Fe0.2−yZryO3−δ (y ≤ 0.10) powders for (i) in situ phase evaluation by X-ray diffraction (XRD), (ii) determining oxidation states of species with relative amounts by X-ray photoelectron spectroscopy (XPS), and (iii) membrane making to get better insight about the material (with zirconium) in terms of oxygen permeability, phase stability, and continuous real operation at 900 °C.
Experimental
SrCo0.8Fe0.2−yZryO3−δ (y = 0. 0.025, 0.050, 0.075, 0.10) powders were prepared by sol-gel technique using appropriate mol % of anhydrous strontium nitrate (Sr(NO3)2) (Qualigens), cobalt nitrate hexahydrate (Co(NO3)2·6H2O) (Loba), ferric nitrate nano-hydrate (Fe(NO3)3·9H2O) (Qualigens), zirconyl nitrate hydrate (ZrO(NO3)2·H2O) (Loba), and oxalic acid dihydrate (C2H2O4·2H2O) (Rankem) as precursors. Ethyl alcohol/ethanol (C2H5OH) (Hayman) and distilled water were used as solvent. First, each precursor was separately dissolved in ~ 200 ml of ethanol at 35–40 °C under vigorous stirring for 30 min to get a transparent solution. Due to poor solubility of strontium nitrate and zirconyl nitrate in ethanol, approximately 5 ml of distilled water was added dropwise to achieve complete dissolution. Second, the nitrate salt solutions were poured in a tray and mixed thoroughly before adding oxalic acid solution gradually and the mixture was stirred continuously to form a dark orange gel. Third, the product was digested (i.e., left as such for reaction to occur) for 12 h, dried at 80 °C for 24 h, then ground using an agate mortar-pestle, and finally sieved to obtain a fine light almond color powder (presumably of oxalate; evident from a strong peak visible at 1640 cm−1 in its Fourier transform infra-red (FT-IR) spectrum) [33]. The dried sol-gel oxalate was finally decomposed at 950, 1150, and 1300 °C each for 5 h by raising the temperature at a rate of 3 °C/min. The product was then furnace cooled to room temperature. The resulting black SrCo0.8Fe0.2−yZryO3−δ (y = 0–0.10) powders were subjected to grinding and sieving (through a 240 mesh) for studying their morphology, structural stability, etc. For oxygen permeation measurements, pellets were made by pressing the powder in a die (size ~ 10-mm diameter) at a hydraulic pressure of 50 kN, then annealed at 1300 °C (by raising the temperature at a rate of 3 °C/min) for 5 h and cooled in furnace itself to room temperature. The pellets/discs/membranes (diameter ~ 10 mm and thickness 1–2 mm) so obtained correspond to relative density of better than 95% of the bulk value.
An X-ray diffractometer (PANalytical X’Pert PRO) with CuKα1 radiation (wavelength λ = 1.540598 Ǻ) was engaged for identification of phases present and evaluation of their stability in the temperature range 27–1000 °C. A field emission scanning electron microscope (Carl Zeiss NTS GmbH- SUPRA 40VP) was used for observing the morphology of dried sol-gel products (presumably oxalates), oxide powders, and membranes. An X-ray photoelectron spectrometer (PHI 5000 Versa Probe, ULVAC- PHI) using AlKα (1486.6 eV) radiation was employed for ascertaining the oxidation states of iron and cobalt species. A laboratory reactor, combined with a gas chromatograph (NUCON, Model 5765) and a molecular sieve 5A capillary column, was employed for measuring the oxygen permeability of membranes in the temperature range of 700–1000 °C using air as feeding gas (400 ml/min) and helium as a carrier (45 ml/min). A fine alumina (size ~ 50 nm) paste–coated inconel washer served as sealant for SrCo0.8Fe0.2−yZryO3−δ (y = 0–0.10) membranes. The alumina paste was also applied over the side curved surface of the disc membrane to prevent radial leakage of oxygen in the reactor. The nitrogen content of the collected gas was simultaneously measured to allow correction in data for air leakage through pore and/or around the membrane. The membrane was maintained at each temperature for about 30–45 min to attain equilibrium before noting down the permeation reading.
Results and discussion
Microstructure and phases
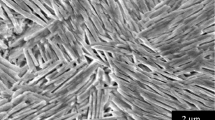
The powder samples displayed different morphologies depending on the composition (Fig. 1). For example, numerous sticks lying parallel to each other in different groups (orienting randomly) are visible with some overlap at places−giving appearance of a thatched roof, mat, or nest of twigs in composition y = 0 (Fig. 1a). On the other hand, samples looked like algae or leaf for y = 0.025 and 0.05 (Fig. 1b, c), islands/regions with gaps for y = 0.075 (Fig. 1d), and bars/parallelepiped for y = 0.10 (Fig. 1e).

Scanning electron micrographs (SE mode) of sol-gel products after drying at 80 °C for 24 h to yield SrCo0.8Fe0.2−yZryO3−δ with y equal to a 0, b 0.025, c 0.050, d 0.075, and e 0.100 following decomposition at elevated temperatures
Figure 2 shows the weight (W%) and dW(%)/dT versus temperature plots of dried sol-gel powder, conforming to composition SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.05, and 0.10), obtained by raising the temperature at a rate of 4 °C per minute up to 900 °C in air. Three main stages of weight loss found correspond to removal of (i) water, (ii) crystallizing water and carbon monoxide, and (iii) residual CO and CO2. The decomposition was nearly complete at ~ 900 °C with a total loss of about 54–57% for the compositions y = 0, 0.05, and 0.10. The presence of zirconium causes less reduction in weight above 300 °C but leads to abrupt significant loss in the temperature range of 600–665 °C. It means that volatile species are retained by strong bonds and released eventually above 600 °C. This difference perhaps facilitates the formation of algae/leaf or other morphology observed in oxalate powder containing zirconium.

Weight percent (W%) and d(W%)/dT versus temperature (T) plots of dried sol-gel product, synthesized for the preparation of SrCo0.8Fe0.2−yZryO3−δ with y = 0, 0.05, and 0.10
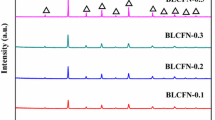
Figure 3 shows the XRD patterns of SrCo0.8Fe0.2−yZryO3−δ powders, obtained by decomposition of dried sol-gel oxalate product at 1150°or 1300 °C for 5 h in air. The diffraction patterns (Fig. 3a, b) of compositions y = 0 and 0.025 correspond to a single phase and can be indexed based on perovskite-type cubic structure with lattice parameter a = 3.8650 and 3.8667 ± 0.0002 Å, respectively. Figure 3a matches well with SrCo0.8Fe0.2O3−δ (JCPDS File no. 82-2445). At higher zirconium content (y = 0.050, 0.075, and 0.100), XRD contain some extra peaks (marked by asterisk (*) in Fig. 3) belonging to a secondary SrZrO3 phase (JCPDS file no. 76-0167), which remain present even after sintering at 1300 °C for 5 h (Fig. 3b). This finding suggests that the solubility limit of zirconium in SrCo0.8Fe0.2ZryO3−δ conforms to y ≤ 0.05. The formation of SrZrO3 has been observed earlier in SrCo0.4Fe0.6−xZrxO3−δ (x = 0.15, 0.2), Sr(Co0.8Fe0.2)1−xZrxO3−δ (x = 0.06), Sr(Co0.9Zr0.1)O3−δ, and SrCo0.4Fe0.5Zr0.1O3−δ systems as well [22, 27, 34, 35]. The analysis of XRD patterns reveal progressive increase in lattice parameter of perovskite-type SrCo0.8Fe0.2−yZryO3−δ cubic phase with rise in zirconium content (Fig. 3b); the values being 3.8650, 3.8667, 3.8690, 3.8758, and 3.8805 Å (± 0.0002 Å) for samples of compositions y = 0, 0.025, 0.050, 0.075, and 0.100, respectively. The increase in parameter (by ~ 0.4% maximum) may be attributed to partial substitution of iron ions with large zirconium species (rFe4+ = 0.585 Å, rFe3+ = 0.55 Å (low spin) or 0.645 Å (high spin), rZr4+ = 0.72 Å). Obviously, zirconium insertion causes gradual expansion, increases the volume of SrCo0.8Fe0.2−yZryO3−δ cubic unit cell, and thus makes the structure somewhat open. The average crystallite size (D) of powder samples as deduced from the Scherrer formula (D = 0.9 x λ/(β cos θ), where λ is the wavelength of X-rays, β is the corrected full width at half maximum (FWHM), and θ is the Bragg angle), taking silicon as standard and diffraction data of 110 peak, turns out to be ~ 100 ± 2 nm for samples having no zirconium and in the range (79–118) ± 2 nm after zirconium addition. While the decrease in size is caused by impurity effect (addition of zirconium), the increase in D occurs due to grain growth brought by sintering at the elevated temperature (1300 °C), used for synthesis of compositions y = 0.075 and 0.100. D values of SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.050, 0.075, and 0.010) powder samples are 100, 79, 83, 100, and 118 nm, respectively. Figure 4a displays a typical scanning electron micrograph of SrCo0.8Fe0.2O3−δ powder sample obtained by decomposition of sol-gel derived oxalate at 1150° for 5 h. The corresponding zoom image shown in Fig. 4b indicates the presence of agglomerates of small crystallites. Similar morphology with no apparent variation is observed in samples containing zirconium.

X-ray diffraction patterns of SrCo0.8Fe0.2−yZryO3−δ obtained from dried sol-gel oxalate product after decomposition for 5 h each at (a) 1150 °C for y = 0, 0.025, 0.050, 0.075, and 0.010 and (b) 1300 °C for y = 0.075, and 0.100. The 110, 200, and 211 peaks of cubic SrZrO3 (a = 4.094 Å) are marked by asterisk (*)

Scanning electron micrographs (SE mode) of a SrCo0.8Fe0.2O3−δ powder and its b zoom version of a lump showing agglomeration of small crystallites
Temperature induced phases
Figure 5a shows a few selected XRD patterns of a SrCo0.8Fe0.2O3−δ sample, recorded at different temperatures in the range of 27–1000 °C. As discussed above, the pattern at 27 °C corresponds to a perovskite-type cubic unit cell with lattice parameter a = 3.8650 ± 0.0002 Å. With increase in temperature, XRD pattern changes and the system appears transforming gradually to an orthorhombic phase assuming lattice parameters ao ~ a√2, bo ~ 4a, and co ~ a√2 (where “a” corresponds to the size of the primary SrCo0.8Fe0.2O3−δ cubic unit cell) [18, 36,37,38,39]. Incidentally, this phase matches with the known brownmillerite (Ca2AlFeO5)-type orthorhombic structure having lattice parameters ao = 5.5981(3) Å, bo = 15.7832(8) Å, co = 5.4692(3) Å, Z = 4, and space group Ibmm (JCPDF no. 82-2446), which supposedly have ordering of anionic vacancies. It can be described by composition A2+B3+O2.5 (A = Ca2+, B = Al3+, Fe3+) with BO6 octahedra and BO4 tetrahedra in alternating layers along the b-axis [40]. The crystal structure of a similar compound Ba2InGaO5 has been studied in detail by Didier et al. [41] recently. Accordingly, it displays a brownmillerite orthorhombic structure with cell parameters a = 6.11239(4) Å, b = 15.56492 (2) Å, and c = 5.92212(3) Å, Z = 4, and space group Ibm2 at 1100 °C. Indium and gallium ions are ordered and occupy octahedral and tetrahedral sites, respectively, because of their size difference (radii being rIn = 0.80 Å, rGa = 0.62 Å). A transition occurs at 1300 °C to a stable perovskite cubic structure assuming a = 4.1539(9) Å, whereas the mixture of the two phases is observed in the intermediate temperature range (1100–1300 °C). The cubic phase depicts disordering of B-cations (In, Ga) as well as anions in Ba2In2O5 and ascribed to high ionic conductivity [42]. Also, the above orthorhombic-cubic transition is reversible in nature. The split in the diffraction peak 110 of perovskite-type cubic is, in fact, indicative of the presence of a brownmillerite-type orthorhombic phase with oxygen vacancy ordering in SrCo0.4Fe0.6O3−δ system [23]. Above 800 °C, a stable single perovskite-type cubic phase [17, 39] with increased lattice parameter (a = 3.9937 ± 0.0002 Å) emerges at 1000 °C. The increase in “a” by ~ 3.3% can be ascribed to thermal expansion. Further, electrons released with oxygen desorption as molecules above 800 °C contribute in part for B4+ → B3+ conversion of Co and/or Fe species and, in turn, the B–O bond length stretches (i.e., increases) further. The weak B-O bonding relaxes the lattice strain and facilitates orthorhombic–cubic transformation besides disorder of oxygen vacancies perhaps. The weak B-O bonding relaxes the lattice strainand facilitates orthorhombic–cubic transformation besides disorder of oxygen vacancies perhaps. Since the orthorhombic structure has an ordered distribution of anion vacancies, movement of oxygen ions in that phase is somewhat restricted amounting to poor oxygen permeability [38]. The structural changes above 800 °C involve oxygen evolution with order → disorder transition of anion vacancies. Such a state is expected to facilitate oxygen flow in the new phase emerging above 800 °C with an enlarged perovskite-type cubic cell.


a XRD patterns of a typical SrCo0.8Fe0.2O3−δ (SCFO) powder sample at different temperatures in the range of 27°–1000 °C. b Summary of phases present in SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.05, and 0.1) samples at different temperatures in the range of 27°–1000 °C
XRD diffraction patterns of SrCo0.8Fe0.2−yZryO3−δ (y = 0.025–0.100) powders recorded in the temperature range of 27–1000 °C reveal interesting effects of zirconium, which can be summarized as under (Fig. 5b):
- a)
The temperature of cubic → brownmillerite orthorhombic-phase transition increases from 200 to 500 °C for the composition y = 0.025. However, for the case of SrCo0.8Fe0.2−yZryO3−δ (y = 0.050), the orthorhombic phase emerges at 200 °C itself (with little SrZrO3) and remains stable up to 800 °C when it transforms back to cubic with enlarged unit cell, of course.
- b)
At zirconium content (y = 0.075 and 0.100), an added quantity of SrZrO3 precipitates out and the cubic → orthorhombic transition occurs at 700 °C or above with the system becoming strontium deficient.
- c)
The orthorhombic phase exists in SrCo0.8Fe0.2−yZryO3−δ system above 500, 700, and 750 °C for compositions y = 0.025, 0.075, and 0.100, respectively.
- d)
An enlarged stable perovskite cubic phase appears above 800 °C for the zirconium content y = 0 and 0.05.
The partial substitution of iron by zirconium in SrCo0.8Fe0.2O3−δ modifies the average oxidation state of Fe/Co and, in turn, reduces the oxygen loss (δ). At y = 0.050, zirconium dissolution in SrCo0.8Fe0.2O3−δ induces enough structural distortion due to its large size and facilitates cubic → orthorhombic transformation just above 100 °C. However, upon increasing zirconium content beyond y = 0.050, SrZrO3 precipitates out with depletion of strontium and release of stress, if any. Consequently, initial perovskite cubic phase remains stable and transforms to orthorhombic phase only above 700–750 °C.
X-ray photoelectron spectroscopy studies
Figure 6 shows the XPS spectrum of SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.050), obtained with a PHI 5000 Versa Probe photoelectron spectrometer model ULVAC using AlKα radiation (1486.6 eV) under vacuum of better than 6.7 × 10−8 Pa at a low resolution (step 1 eV) in the energy range of 0–900 eV. The peaks are identified as belonging to Sr3d, C 1 s + Sr3p1/2, Co2p, Fe2p, O1s, and Zr3d levels. The carbon 1 s peak at 284.6 eV is treated as standard for calibration. The corresponding fine spectra were observed in different energy regimes at a high resolution (step 125 meV). The de-convoluted Gaussian peaks fitted using Origin 8.5 software for cobalt, iron, and oxygen are shown in Fig. 7. The values of binding energy for different species in SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.050) system are summarized in Table 1. Their nature can now be discussed separately:
- (a)
Co 2p

XPS survey spectra of SrCo0.8Fe0.2−yZryO3−δ with y = (a) 0, (b) 0.025, and (c) 0.050

XPS spectra showing Gaussian fitting of peaks for (a1, a2, a3) Co 2p, (b1, b2, b3) Fe 2p, and (c1, c2, c3) O 1 s in SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.050) system
Figure 7(a1) depicts a high-resolution XPS scan of SrCo0.8Fe0.2O3−δ in the energy range of 771–816 eV with two peaks of Co 2p3/2 and Co 2p1/2 levels. Dupin et al. [47] have described the XPS spectra of cobalt in Li0.66CoO2 and LiCoO2 systems in detail. Accordingly, Co 2p peak appearing in the energy range 775–800 eV is built up of signals from cobalt (3 +) and (4 +) states. While a doublet with energies 779.8 and 795 eV corresponds to Co3+ ions, the other doublet with peaks at 781.5 and 796.8 eV belongs to Co4+ ions. In the present case, de-convolution exercise of XPS spectrum presented in Fig. 7(a1) reveals two doublets with binding energies (779.8 and 795.1 eV) and (781.4 and 796.7 eV). They can therefore be ascribed to Co3+ and Co4+ ions, respectively. The peak position and (%) peak area of each oxidation state as deduced from Co 2p3/2 peaks (Fig. 7(a1, a2,a3)) are listed in Table 2.The relative amounts of Co3+ and Co4+ ions, given by respective peak areas are 55.9% and 44.1%, respectively, in the case of SrCo0.8Fe0.2O3−δ system.
- (b)
Fe 2p
Figure 7(b1) shows a high-resolution XPS scan of SrCo0.8Fe0.2O3-δ in the energy range 701–741 eV and contains information about the oxidation states of iron. The peak broadness at first sight suggests the presence of all three Fe3+, Fe2+, and Fe4+ions, arising due to electrostatic interactions, spin-orbit coupling between the 2p core hole and unpaired 3d electrons of the photo-ionized cation, and crystal field effects [48, 49]. Yunfei et al. [46] have classified peaks of Fe 2p3/2 level in the energy range of 709.6–709.9 eV, 710.9–711.9 eV, and 712–714 eV for Fe2+, Fe3+, and Fe4+species, respectively. The Gaussian fitting of Fe 2p shown in Fig. 7(b1) corresponds to two peaks with binding energy 710.9 and 714.2 eV, which can be attributed to Fe3+ and Fe4+ ions, respectively (Table 2). The peak position, % peak area, and oxidation state obtained from Fe 2p3/2 peaks (Fig. 7(b1, b2, b3)) are summarized in Table 2. Note that the Fe3+ ions present in SrCo0.8Fe0.2O3-δ system is just 8.5% while the Fe4+ species share the remaining 91.5%.
- (c)
O 1 s
The high-resolution XPS spectrum in the energy range 525–543 eV depicted in Fig. 7(c1) gives the signature of oxygen ions. The Gaussian fitting of peak shows splitting and associated binding energies are 529.5 and 531.3 eV (Fig. 7(c1)). Machocki et al. [50] identified two peaks for oxygen in the XPS spectrum of manganese-lanthanum oxides: (i) at lower binding energy (529.4–529.8 eV) belonging to oxygen ion (O2−) and (ii) slightly higher energy range of 531.2–531.8 eV arising due to O 1 s states of surface adsorbed oxygen (O22−, O2−, or O−). Thus, peaks of O 1 s observed at 529.5 and 531.3 eV can be assigned to oxygen ion (O2−) and surface adsorbed oxygen, respectively. The peak position, % peak area, and nature of oxygen resulting from O 1 s peaks (Fig. 7(c1, c2, c3)) in SrCo0.8Fe0.2O3-δ are given in Table 2. These indicate that the surface adsorbed oxygen (O22−, O2−, or O−) amounts to 91.4% whereas the bulk oxygen ion (O2−) is just 8.6%.
- (d)
Zr 3d
Zhang et al. [44] have resolved the Zr 3d spectrum and assigned binding energies 184.4 eV and 182.0 eV to Zr 3d3/2 and Zr 3d5/2, respectively, in close agreement with the stoichiometric ZrO2 data. Accordingly, XPS peaks observed in Fig. 6b at 183.1 and 180.8 eV correspond to Zr 3dp3/2 and Zr 3d5/2 level, respectively, for SrCo0.8Fe0.2−yZryO3−δ (y = 0.025). These peaks shift to higher values 183.3 eV and 181.1 eV, respectively, when the zirconium content is increased to y = 0.05.
The relative amounts of cobalt and iron species in 3 + and 4 + oxidation states deduced from XPS data of SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.050) are summarized in Table 3. These clearly point out that zirconium addition induces steady (i) decrease in Co3+ and Fe4+ species and (ii) increase in Co4+ and Fe3+ ions in SrCo0.8Fe0.2−yZryO3−δ (y = 0, 0.025, 0.050). Thus, zirconium (which is in 4 + oxidation state) replaces Fe4+ species in SrCo0.8Fe0.2O3−δ and reduces their number. In addition, there is (i) proportionate Co3+ → Co4+change accompanied by release of electrons and (ii) some Fe4+ → Fe3+conversion with consumption of electrons. The net result is an effective increase in species of 4 + oxidation state with rise in zirconium amount (Table 3). There should therefore be a corresponding increase in oxygen ions (or decrease in anion vacancies, i.e., reduction in δ) for ensuring charge neutrality(discussed in the “X-ray photoelectron spectroscopy studies” section). The oxygen gain by the material can account for the changes in the oxidation states of species. The additional oxygen molecules adsorb turned into anions by consuming electrons released via Co3+ → Co4+ conversion. The oxygen gain is evident from the XPS analysis given in Table 2 as O2− content is increasing progressively with rise in zirconium content y (= 0.025 and 0.050) in SrCo0.8Fe0.2−yZryO3−δ.
Oxygen deficiency parameter (δ)
The oxygen deficiency parameter (δ) in SrCo0.8Fe0.2−yZryO3−δ can be estimated with the principle of charge balance, taking the oxidation states of iron and cobalt as 3 + and/or 4 +, and by considering variation in their contents. The structural formula can therefore be rewritten as Sr2+Co3+0.8−xCo4+xFe3+0.2−y−zFe4+zZr4+yO2−3−δ with δ = (1−x−y−z)/2, where x = (0–0.8), y = (0–0.1), and z = (0–0.2) denote Co4+, Zr4+, and Fe4+ content, respectively. In the ideal case, δ is zero where A and B sites in perovskite cubic structure (ABO3) assume oxidation state 2 + and 4 +, respectively. However, if B species display both 3 + and 4 + states, δ becomes non-zero, indicating presence of oxygen vacancies. The value of “δ” lies in the range 0–0.5. Notice that δ, besides Zr4+ content, depends on the relative amounts of (Fe3+, Fe4+) and (Co3+, Co4+). XPS analysis was used to estimate the relative amounts of oxidation states of iron and cobalt species lying on the surface of the membranes. According to Table 2, SrCo0.8Fe0.2O3−δ contains cobalt and iron ions in oxidation state (4 +) and (3 +) with oxygen species (O22−, O2−, or O−). The contents of species as found are Co4+ (44.11%), Co3+ (55.89%), Fe4+ (91.5%), Fe3+ (8.5%), surface adsorbed oxygen O22−, O2−, or O− (91.4%), and bulk oxygen O2− (8.6%). Zirconium incorporation at Co/Fe sites leads to increase of Co4+, Fe3+, and O2− ions while decrease of Co3+, and Fe4+ ions. The net effect amounts to increase of total positive charge as well as number of oxygen O2− ions (i.e., lowering of δ). Considering the relative amounts of Co3+, Co4+, Fe3+, and Fe4+ present, oxygen deficiency parameter (δ) has been deduced by invoking the concept of charge neutrality. This exercise gives the value of δ as 0.23, 0.22, and 0.21 for SrCo0.8Fe0.2−yZryO3−δ with y = 0, 0.025, and 0.050, respectively. Obviously, there is minor decrease in oxygen deficiency parameter δ (or increase in anion concentration) with the increase in zirconium content. The above figures no doubt pertain to sample surface at room temperature, yet very useful in understanding the phenomenon at high temperatures.
Oxygen permeability
The oxygen permeation flux (JO2) is specified in units of milliliters per square centimeter per minute or moles per square centimeter per second; the conversion relation being 1 ml cm−2 min−1 = 7.44 × 10−7 mol cm−2 s−1. JO2 was measured in SrCo0.8Fe0.2−y ZryO3−δ (y = 0–0.1) membrane discs (diameter ~ 10 mm and thickness ~ 1.0–2.0 mm) at 700–1000 °C with air as a feeder gas on the front (or input) and helium as a carrier on the exit (or sweep) side [51]. Figure 8a depicts (JO2) versus temperature plots for SrCo0.8Fe0.2O3−δ membranes (thickness 1.0, 1.5, and 2.0 mm; effective diameter 8.8 mm), obtained using input air and sweeping helium gas flow rates of 400 ml/min and 45 ml/min, respectively. It may be noticed that JO2 increases with temperature invariably and is more for a thin membrane, and attains a maximum value of ~ 2.44 ml/cm2·min (or 1.8 × 10−6 mol cm−2 s−1) at 1000 °C for 1.0 mm-thick disc. The corresponding ln (JO2) vs. 1000/T plots are shown in Fig. 8b. They are nearly linear and follow the Arrhenius equation JO2 = JO exp (− EA/kBT), where Jo is the pre-exponential factor, EA is the activation energy of oxygen permeation, and kB is the Boltzmann constant. The values of activation energy, as deduced from the slopes of linear portions in the temperature range 800–1000 °C, are 50.0, 52.4, and 56.2 kJ/mol (or 0.52, 0.54, and 0.58 eV/ion) for membranes of thickness 1.0, 1.5, and 2.0 mm, respectively. The increase in EA is in conformity with the observed decrease in the oxygen permeation flux for thicker membranes.

a Oxygen permeation flux (JO2) versus temperature and b ln (JO2) vs. 1000/T Arrhenius plots of SrCo0.80Fe0.20O3−δ membranes of thickness 1.0, 1.5, and 2.0 mm (diameter of each being ~ 10 mm)
Earlier, SrCo0.8Fe0.2O3−δ membranes were fabricated with powders derived from liquid citrate and solid-state reaction routes. The maximum oxygen permeability flux (JO2) of 1-mm-thick membrane was found to be 1.8 × 10−7 or 6.3 × 10−7 mol cm−2 s−1 at 850 °C [17, 18]. However, Teraoka et al. [11] reported a value of 2.31 × 10−6 mol cm−2 s−1 (i.e., an order higher) in this system under similar operating conditions. The reason for this discrepancy is not apparent and remains still unanswered. It is another matter that significant variation in JO2 is usually observed even in identical membranes [29]. In the present study, JO2 value of SrCo0.8Fe0.2O3−δ membrane (thickness 1.0 mm), fabricated using sol-gel-based powder, is slightly higher ~ 9.9 × 10−7 mol cm−2 s−1 (or 1.33 ml cm−2 min−1) at 850 °C (with input air flow rate of 400 ml/min and sweeping helium flow rate of 45 ml/min) than those reported by Kruidhof et al. [17] and Qiu et al. [18], respectively. The improvement in JO2 has origin with high purity powder and a mechanically stable membrane [31, 52]. It is due to homogeneous powder of fine particles and, in turn, a stable compact disc of high density (> 95%) obtained via sol-gel route (preferred here over solid-state reaction), which ensures steady and full mixing of precursor species at the atomic/ molecular level and provides better stoichiometry control.
The processes occurring at front and exit surfaces and within the membrane disc are illustrated schematically in Fig. 9. A driving force for oxygen permeation is the partial pressure gradient prevailing along the membrane thickness. The main steps involved in permeation process are:
- (a)
selective adsorption of oxygen molecules on the front surface (at high pressure) and their dissociation into ions by capturing electrons following the reaction O2 + 4e− → 2O2−,
- (b)
migration/diffusion of oxygen ions through the membrane thickness via anion vacancies to the exit (sweeping) side,
- (c)
conversion of oxygen ions into molecules by leaving behind the electrons before desorbing at the exit surface, the reaction being 2O2− → O2 + 4e−, and
- (d)
movement of electrons backwards from the exit to the front side of the membrane due to concentration gradient and/or via hopping following the scheme

Schematic diagram illustrating processes occurring at front and exit surface, oxygen transport, and electron movement in a typical SrCo0.8Fe0.2O3−δ membrane
At elevated temperatures, JO2 increases due to cumulative effects of (i) additional anion vacancy generation, (ii) enhanced surface reactions/improved dissociation of oxygen molecules, and (iii) easy forward migration of O2− ions and backward movement of electron within the membrane.
The partial replacement of B species by zirconium (Zr) in SrCo0.8Fe0.2O3−δ stabilizes the perovskite-type cubic structure and induces disordering in oxygen vacancies as well [22, 23]. Also, increased oxygen permeability of La0.2Ba0.8Co0.8Fe0.2−xZrxO3−δ (x = 0.05) at elevated temperatures is attributed to prevailing disorder in anion vacancies [24]. Zirconium in BaCo0.7Fe0.3−xZrxO3−δ (x = 0–0.12) increases JO2 up to x = 0.06 but decreases at higher content [25]. Further, ZrO2 containing SrCo0.4Fe0.6O3−δ membrane not only depicts better oxygen permeability (JO2) but also remains stable at elevated temperatures and under reduced oxygen partial pressures [26]. On the contrary, Yang et al. [22, 23] observed improved phase stability in SrCo0.4Fe0.6−xZrxO3−δ (x = 0–0.2) membranes at the cost of perceptible decrease in oxygen permeability. Chen et al. [27] and Lu et al. [28] also reported similar results (i.e., improved structural stability and reduced oxygen permeability) in Zr-doped SrCo0.8Fe0.2O3−δ and Ba0.5Sr0.5Co0.8Fe0.2O3−δ membranes, respectively. Obviously, zirconium incorporation in SrCo0.8Fe0.2O3−δ compound has led to conflicting oxygen permeability data. The stability of perovskite structure is also known to improve with addition of fixed valence state elements [27]. Further, increase in unit cell size (i.e., realizing a somewhat open structure) may facilitate movement of anions and enhance oxygen permeability. With zirconium, oxygen vacancy concentration should decrease due to its oxidation state (4 +) and is reflected in room temperature XPS data as well. However, membrane operates at elevated temperatures where some oxygen evolution also occurs and compensates for the loss.
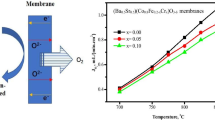
The effect of zirconium on oxygen permeation of SrCo0.8Fe0.2−yZryO3−δ (y = 0–0.10) membranes (thickness 1, 1.5, and 2 mm) at 700–1000 °C is shown in Fig. 10. The measurements were repeated 3–4 times in each case for consistency and data reliability. Accordingly, zirconium insertion enhances the oxygen permeability initially (up to y = 0.05) but reduces at higher concentrations at all temperatures (Table 4). As for example, 1.5-mm-thick membrane at 1000 °C reveals increase in JO2 to 2.91 ml/cm2·min for the Zr content y = 0.05 but exhibits a lower value of 1.66 ml/cm2·min for y = 0.10. Notice that JO2 value for 1-mm-thick membrane at 1000 °C advances from 2.44 ml/cm2·min to 3.42 ml/cm2·min when zirconium content (y) increases from 0 to 0.05. Thus, 5% zirconium at B site (initially occupied by cobalt and iron species together or 25% replacement of iron alone) surges the oxygen permeability by about 40%. This happens due to (i) a stable perovskite-type cubic structure with random distribution of anion vacancies at elevated temperatures, (ii) enlarged unit cell because of bigger Zr4+ ion insertion and thermal expansion, (iii) impending increase in O2− vacancies following desorption of oxygen as molecules with simultaneous release of electrons, and (iv) enhanced surface reactions, i.e., improved dissociation of oxygen molecules at the front surface. At higher zirconium content (y ≥ 0.05), the lower oxygen permeation capacity results because of (i) stabilization of the brownmillerite orthorhombic structure and (ii) emergence of SrZrO3 as a secondary phase. These findings suggest that the dissolution limit of zirconium in SrCo0.8Fe0.2−yZryO3−δ lies below y = 0.05. It may be recalled that the above observation is consistent with that displayed by La0.2Ba0.8Co0.8Fe0.2−yZryO3−δ membrane (thickness 1 mm), i.e., enhancement of oxygen permeability upon raising y from 0 to 0.05; the values at 1000 °C being 1.1 and 2.69 ml/cm2·min, respectively. Fan et al. [19] attributed increased oxygen permeation at 700–1000 °C in the perovskite cubic structure to random distribution of anion vacancies.

Oxygen permeation flux (JO2) versus temperature plots of SrCo0.80Fe0.20−yZryO3−δ membranes of diameter ~ 10 mm and thickness a 1.0 mm, b 1.5 mm, and c 2.0 mm
The oxygen permeability for all membranes is deduced for completeness and to demonstrate its inverse thickness dependence. JO2 in membrane (thickness > 0.3 mm) is governed by diffusion-controlled process and expressed at a temperature (T) as [9, 29].
where R is the gas constant, σO (PO)−n is the conductivity (σ; also proportional to anion vacancy concentration), F is flow rate of carrier gas, L is the membrane thickness, and PO stands for the oxygen pressure (subscripts I and E representing inlet and exit side) with n as an exponent. Notice decrease in JO2 with increase in thickness (Figs. 8 and 10). Also, the objective has been to infer the optimum thickness displaying realistic JO2 with high mechanical stability. The results suggest that 1-mm-thick membrane conform to both these features/characteristics.
SrCo0.8Fe0.2−yZryO3−δ (y = 0.05) membrane corresponds to highest JO2 because of the zirconium (oxidation state 4 + and ionic radius 0.72 Å, larger than Fe4+ and Fe3+) amount being just appropriate for inducing stability to the perovskite-type cubic phase besides creating random distribution of anion vacancies at elevated temperatures (≥ 800 °C) [22, 23, 27]. At other concentrations, the system shows (Fig. 5b) either the brownmillerite orthorhombic phase (Sr-deficient) with SrZrO3 precipitation above 700–750 °C (for y = 0.075, 0.010) or a lone orthorhombic phase above 500 °C (for y = 0.025). In both the cases, JO2 values are lower than displayed by cubic phase of compositions y = 0, 0.05 at ≥ 800 °C (Fig. 10). But, SrCo0.8Fe0.2O3−δ (i.e., with no zirconium content y = 0) depicts lower JO2 and poor long-term stability vis-à-vis the compound having y = 0.05 (Figs. 10 and 12).
Ln (JO2) vs. 1000/T plots for SrCo0.80Fe0.20−yZryO3−δ (y = 0–0.10) membranes of thickness 1.0, 1.5, and 2.0 mm display linearity in the temperature range of 800–1000 °C. The activation energy obtained from the slope reveal decrease first but increase beyond zirconium concentration of y = 0.05. For example, EA values of 1.5-mm-thick membrane found are 52.4, 50.3, 54.6, and 55 kJ/mol for y = 0, 0.05, 0.075, and 0.100, respectively. The decrease (or increase) in the activation energy is compatible with the increase (or decrease) observed in JO2. This feature may be related to limited solubility of zirconium in SrCo0.80Fe0.20O3−δ and precipitation of SrZrO3 above a critical value of y ~ 0.05.
Membrane stability and reliability
To ascertain the structural stability of SrCo0.8Fe0.2O3−δ membrane, XRD patterns of both the front and exit surfaces were recorded and analyzed after completion of three cycles of oxygen permeation measurements at 700, 800, 900, and 1000 °C in succession. XRD patterns of 1-mm-thick membrane depicted in Fig. 11 reveal clearly the existence of only perovskite-type cubic phase on both sides after three oxygen permeation cycles at 700–1000 °C covering a total time of about 24 h. Obviously, the SrCo0.8Fe0.2O3−δ disc remains stable all through the operation cycles. Nevertheless, for practical applications, the membrane should be reliable in terms of oxygen permeability as well as long-term stability. To investigate this aspect, (JO2) of SrCo0.8Fe0.2O3−δ membrane (thickness 1.5 mm) was monitored at 900 °C for 120 h through a continuous operation by setting the flow rates of input air at front surface and carrier helium gas at exit side as 400 ml/min and 45 ml/min, respectively (Fig. 12a). Notice that the JO2 figure was sustained at ~ 1.41 ml/cm2·min for barely 60 h and fell thereafter by ~ 7% during the next 60 h. Undoubtedly, this JO2 value is above 1.0 ml/cm2·min set for economic and industrial viability considerations [53], yet the long-term stability of the membrane remained poor and caused concern. In this context, partial substitution of iron with zirconium in SrCo0.8Fe0.2O3−δ was rather advantageous. For example, the oxygen permeation flux of SrCo0.80Fe0.15Zr0.05O3−δ membrane (thickness 1.5 mm) measured at 900 °C shows a constant higher value of 1.99 ml/cm2·min for 120 h (Fig. 12b). Obviously, zirconium incorporation has not only caused significant improvement in (JO2) but also induced marked stability in the SrCo0.80Fe0.20O3−δ membrane. Also, its XRD patterns before and after carrying out the oxygen permeation exercise at 900 °C for 120 h (shown in Fig. 13) indicate continual presence of perovskite-type cubic phase and so the structural stability of the membrane. Similarly, SrCo0.8Fe0.2O3−δ membrane (thickness 1 mm) exhibits high oxygen permeation flux JO2 of ~ 2.44 ml/cm2·min (or 1.8 × 10−6 mol cm−2 s−1) at 1000 °C but shows inadequate long-term stability. However, zirconium substitution of iron (i) makes the perovskite-type cubic phase stable in the temperature range 800–1000 °C with lattice parameter a = 3.9629–3.9873 Å; (ii) enhances JO2 by ~ 40%, i.e., to ~ 3.42 ml/cm2·min at 1000 °C in SrCo0.8Fe0.2−yZryO3−δ (y = 0.05) membrane of thickness 1 mm (Fig. 10a); and (iii) sustains steady oxygen permeation for a longer duration (for at least 120 h at 900 °C). The increase in oxygen permeation flux can be attributed to (a) enlarged perovskite-type cubic unit cell and (b) additional anion vacancies produced by weakening of metal-oxygen bond (brought via thermal expansion) and gaining energy at elevated temperatures. The significant rise observed in oxygen permeability itself is indicative of extra anion vacancies.

X-ray diffraction patterns of SrCo0.8Fe0.2O3−δ membrane (a) as such or before and (b, c) after oxygen permeation process; (b) front/entrance, and (c) back/exit side

The variation of oxygen permeation flux (JO2) for (a) SrCo0.80Fe0.20O3−δ, (b) SrCo0.80Fe0.20−yZryO3−δ (y = 0.05) membranes (thickness = 1.5 mm) held at 900 °C for 120 h. (unit being 1 ml cm−2 min−1 = 7.44 × 10−7 mol cm−2 s−1)

X-ray diffraction patterns of SrCo0.80Fe0.20−yZryO3−δ (y = 0.05) membrane (a) as such, (b, c) after oxygen permeation at 900 °C for 120 h, (b) front/entrance, and (c) back/exit side. The SrZrO3 peak is marked with asterisk (*)
Conclusions
A single perovskite-type cubic phase of SrCo0.8Fe0.2O3−δ (a = 3.8650 ± 0.0002 Ǻ, Z = 1), produced by decomposition of sol-gel derived oxalate precursor at 1150 °C for 5 h, remains stable in the temperature range 800–1000 °C. It is characterized by a high oxygen permeability flux of JO2 ~ 1.65 ml/cm2·min (or 1.23 × 10−6 mol cm−2 s−1) at 900 °C for 1-mm-thick membrane; JO2 is maintained continuously for ~ 60 h only at 900 °C. The 5% zirconium incorporation at Co/Fe site (i) enlarges the perovskite-type cubic unit cell exhibiting a = 3.9629–3.9873 Å in the temperature range of 800–1000 °C, (ii) raises JO2 to ~ 2.34 ml/cm2·min (or 1.74 × 10−6 mol cm−2 s−1) at 900 °C, and (iii) improves phase stability and sustains oxygen permeation for much longer duration (i.e., 120 h at least). The formation of a perovskite-type cubic phase in Sr–Co–Fe–O system with abundance of anion vacancies is crucial for both the high oxygen permeation and extended operational stability at elevated temperatures (800–1000 °C). This can be achieved by partial substitution of iron with zirconium, optimum composition being SrCo0.8Fe0.2−yZryO3−δ (y = 0.05).
References
Hashim SM, Mohamed AR, Bhatia S (2010) Current status of ceramic-based membranes for oxygen separation from air. Adv Colloid Interf Sci 160:88–100
Teraoka Y, Honbe Y, Ishii J, Furukawa H, Moriguchi I (2002) Catalytic effects in oxygen permeation through mixed-conductive LSCF perovskite membranes. Solid State Ionics 152–153:681–687
Bouwmeester HJM, Burggraaf AJ (1996) Fundamentals of inorganic membrane science and technology. Elsevier, Amsterdam Chapter 10
Hong WK, Choi GM (2010) Oxygen permeation of BSCF membrane with varying thickness and surface coating. J Membr Sci 346:353–360
Shao Z, Xiong G, Cong Y, Yang W (2000) Synthesis and oxygen permeation study of novel perovskite-type BaBixCo0.2Fe0.8−xO3−δceramic membranes. J Membr Sci 164:167–176
Vashook VV, Daroukh MA, Ullmann H (2001) Oxygen ion diffusion in perovskite-type oxides determined by permeation and by relaxation measurements. Ionics. 7:59–66
Zeng P, Shao Z, Liu S, Xu ZP (2009) Influence of M cations on structural, thermal and electrical properties of new oxygen selective membranes based on SrCo0.95M0.05O3−δperovskite. Sep Purif Technol 67:304–311
Li K (2007) Ceramic membrane for separation and reaction. John Wiley, England Chapter 6
Sunarso J, Baumann S, Serra JM et al (2008) Mixed ionic–electronic conducting (MIEC) ceramic-based membranes for oxygen separation. J Membr Sci 320:13–41
Teraoka Y, Nobunaga T, Yamazoe N (1988) Effect of cation substitution on the oxygen semipermeability of perovskite-type oxides. Chem Lett 3:503–506
Teraoka Y, Zhang HM, Furukawa S, Yamazoe N (1985) Oxygen permeation through perovskite-type oxides. Chem Lett:1743–1746
Xinfa D, Heng Z, Weiming L (2008) Preparation and characterization of a perovskite-type mixed conducting SrFe0.6Cu0.3Ti0.1O3−δmembrane for partial oxidation of methane to syngas. Chin J Chem Eng 16(3):411–415
Li Y, Zhao H, Xu N et al (2010) Systematic investigation on structure stability and oxygen permeability of Sr-doped BaCo0.7Fe0.2Nb0.1O3−δceramic membranes. J Membr Sci 362:460–470
Wang W, Cong Y, Yang W (2002) Oxygen permeation study in a tubular Ba0.5Sr0.5Co0.8Fe0.2O3-δ oxygen permeable membrane. J Membr Sci 210:259–271
Bouwmeester HJM, Kruidhof H, Burggraaf AJ (1994) Importance of the surface exchange kinetics as rate limiting step in oxygen permeation through mixed-conducting oxides. Solid State Ionics 72:185–194
McIntosh S, Vente JF, Haije WG, Blank DHA, Bouwmeester HJM (2006) Phase stability and oxygen non-stoichiometry of SrCo0.8Fe0.2O3−δ measured by in situ neutron diffraction. Solid State Ionics 177:833–842
Kruidhof H, Bouwmeester HJM, Doorn RHE, Burggraaf AJ (1993) Influence of order-disorder transitions on oxygen permeability through selected nonstoichiometric perovskite-type oxides. Solid State Ionics 63–65:816–822
Qiu L, Lee TH, Liu LM, Yang YL, Jacobson AJ (1995) Oxygen permeation studies of SrCo0.8Fe0.2O3-δ. Solid State Ionics 76:321–329
Fan CG, Deng ZQ, Zuo YB, Liu W, Chen CS (2004) Preparation and characterization of SrCo0.8Fe0.2O3–δ–SrSnO3 oxygen-permeable composite membrane. Solid State Ionics 166:339–342
Lu H, Cong Y, Yang W (2007) Oxygen permeability and improved stability of a permeable Zr-substituted perovskite membrane for air separation. Mater Sci Eng B 141:55–60
Liu H, Zhu Z, Cong Y, Zhang T, Yang W (2012) Remarkable dependence of electrochemical performance of SrCo0.8Fe0.2O3-δ on A-site on stoichiometry. Phys Chem Chem Phys 14:7234–7239
Yang L, Tan L, Gu X, Jin W, Zhang L, Xu N (2003) A new series of Sr(Co,Fe,Zr)O3−δperovskite-type membrane materials for oxygen permeation. Ind Eng Chem Res 42:2299–2305
Yang L, Gu X, Tan L, Zhang L, Wang C, Xu N (2003) Role of ZrO2 addition on oxygen transport and stability of ZrO2-promoted SrCo0.4Fe0.6O3–δ. Sep Purif Technol 32:301–306
Fan CG, Zuob YB, Li JT, Lu JQ, Chen CS, Bae DS (2007) Highly permeable La0.2Ba0.8Co0.8Fe0.2−xZrxO3−δmembranes for oxygen separation. Sep Purif Technol 55:35–39
Yao W, Cheng H, Zhao H et al (2016) Synthesis, oxygen permeability, and structural stability of BaCo0.7Fe0.3−xZrxO3−δ ceramic membranes. J Membr Sci 504:251–262
Li S, Jin W, Huang P et al (1999) Perovskite-related ZrO2 -doped SrCo0.4Fe0.6O3–δmembrane for oxygen permeation. AICHE J 45(2):276–284
Chen W, Zuo YB, Chen CS, Winnubst AJA (2010) Effect of Zr4+ doping on the oxygen stoichiometry and phase stability of SrCo0.8Fe0.2O3−δ oxygen separation membrane. Solid State Ionics 181:971–975
Lu H, Deng ZQ, Tong J, Yang W (2005) Oxygen permeability and structural stability of Zr-doped oxygen-permeable Ba0.5Sr0.5Co0.8Fe0.2O3–δ membrane. Mater Lett 59:2285–2288
Qi X, Lin YS, Swartz SL (2000) Electric transport and oxygen permeation properties of lanthanum cobaltite membranes synthesized by different methods. Ind Eng Chem Res 39:646–653
Martynczuk J, Arnold M, Feldhoff A (2008) Influence of grain size on the oxygen permeation performance of perovskite-type (Ba0.5Sr0.5)(Fe0.8Zn0.2)O3−δmembranes. J Membr Sci 322:375–382
Jaiswal SK, Kashyap VK, Kumar J (2012) On the sol–gel synthesis and characterization of strontium ferrite ceramic material. Mater Res Bull 47:692–699
Kashyap VK, Jaiswal SK, Kumar J (2016) On the structural stability and oxygen permeation behaviorof inorganic SrCo0.8Fe0.2O3−δ membranes. Ionics. 22:2471–2485
Kumar A, Kumar J (2008) On the synthesis and optical absorption studies of nano-size magnesium oxide powder. J Phys Chem Solids 69:2764–2772
Nagai T, Ito W, Sakon T (2007) Relationship between cation substitution and stability of perovskite structure in SrCoO3–δ-based mixed conductors. Solid State Ionics 177:3433–3444
Wu Z, Dong X, Jin W, Fan Y, Xu N (2007) A dense oxygen separation membrane deriving from nanosized mixed conducting oxide. J Membr Sci 291:172–179
Deng ZQ, Yang WS, Liu W, Chen CS (2006) Relationship between transport properties and phase transformations in mixed-conducting oxides. J Solid State Chem 179:362–369
He Y, Zhu X, Guo Z, Yang W (2010) Phase transitions in Sr1+xCo0.8Fe0.2O3−δ oxides. Mater Lett 64:1618–1621
Shin MJ, Yu JH (2012) Oxygen transport of A-site deficient Sr1−xFe0.5Co0.5O3−δ(x = 0–0.3) membranes. J Membr Sci 401–402:40–47
McIntosh S, Vente JF, Haije WG, Blank DHA, Bouwmeester HJM (2006) Structure and oxygen stoichiometry of SrCo0.8Fe0.2O3−δ andBa0.5Sr0.5Co0.8Fe0.2O3−δ. Solid State Ionics 177:1737–1742
Jaiswal SK, Kumar J (2011) On the sol–gel synthesis and structure, optical, magnetic and impedance behaviour of strontium cobaltite powder. J Alloys Compd 509:3859–3865
Didier C, Claridge J, Rosseinsky M (2014) Crystal structure of brownmillerite Ba2InGaO5. J Solid State Chem 218:38–43
Goodenough JB, Ruiz-Diaz JE, Zhen YS (1990) Oxide-ion conduction in Ba2In2O5 and Ba3In2MO8 (M=Ce, Hf, or Zr). Solid State Ionics 44:21–31
Young V, Otagawa T (1985) XPS studies on strontium compounds. Appl Surf Sci 20:228–248
Zhang HL, Wang DZ, Yang B, Huang NK (1997) XPS measurement for the elements in the interface between oxygen ion irradiatedZrO2 – Y2O3 films and iron substrate. Phys Status Solidi 160:145–150
Jiratova K, Mikulova J, Klempa J, Grygar T, Bastl Z, Kovanda F (2009) Modification of Co–Mn–Al mixed oxide with potassium and its effect on deep oxidation of VOC. Appl Catal A 361:106–116
Bu Y, Zhong Q, Xu D, Tan W (2013) Redox stability and sulfur resistance of Sm0.9Sr0.1CrxFe1–xO3–δ perovskite materials. J Alloys Compd 578:60–66
Dupin JC, Gonbeau D, Benqlilou-Moudden H, Vinatier P, Levasseur A (2001) XPS analysis of new lithium cobalt oxide thin-films before and after lithium deintercalation. Thin Solid Films 384:23–32
Norman C, Leach C (2011) In situ high temperature X-ray photoelectron spectroscopy study of barium strontium iron cobalt oxide. J Membr Sci 382:158–165
Grosvenor AP, Kobe BA, Biesinger MC, McIntyre NS (2004) Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf Interface Anal 36:1564–1574
Machocki A, Ioannides T, Stasinska B et al (2004) Manganese–lanthanum oxides modified with silverfor the catalytic combustionof methane. J Catal 227:282–296
Alaee MA, Movahednia MM, Mohammadi T (2009) Effect of Ba content on oxygen permeation performance of BaxSr1-xCo0.8Fe0.2O3-δ(x =0.2, 0.5, and 0.8) perovskite-type membrane. J Chem Eng Data 54:3082–3091
Hench LL, West JK (1990) The sol-gel process. Chem Rev 90:33–72
Steele BCH (1992) Oxygen ion conductors and their technological applications. Mater Sci Eng B13:79–87
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kashyap, V.K., Jaiswal, S.K. & Kumar, J. Effect of Zr4+on the phase stability and oxygen permeation characteristics of SrCo0.8Fe0.2−yZryO3−δ (y ≤ 0.1) membranes. Ionics 26, 1895–1911 (2020). https://doi.org/10.1007/s11581-019-03393-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-019-03393-8