
Abstract
Lithium iron phosphate composite (LiFePO4/C) with uniform carbon coating was synthesized by wet ball-milling, microwave drying, and carbothermal reduction using xylitol-polyvinyl alcohol (PVA) as complex carbon sources. The fused xylitol with the certain viscosity is readily coated on the surface of ferric phosphate (FePO4) during ball-milling. The PVA hydrogel can maintain the precursors stable during the drying process, and the hydrogel also can be transformed into carbon coating around the LiFePO4 during calcination as the additional carbon source. The unique properties of the complex carbon sources result in uniform carbon coating all over the fine spherical particles with an average primary particle size of 350 nm. The particles are connected by a network of filamentous conductive carbon, which provides a channel for Li+ conduction. As a result of this unique structure, the synthesized LiFePO4/C exhibits high electronic and ionic conductivities, which contributes to excellent electrochemical performance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As one of the most promising cathode materials, the olivine-structured LiFePO4 has been widely used in the fields of pure electric vehicles (EVs), plug-in hybrid vehicles (PHEVs), and energy storage systems (ESS) [1,2,3,4]. Since the first reported by Goodenough in 1997, it has been widely studied due to high specific capacity (170 mAh g−1), high thermal stability, low cost, and environmental friendliness [5,6,7,8,9]. However, the main obstacles inhibiting its practical application are low intrinsic electronic conductivity (10−9~10−10 S cm−1) and low Li+ diffusion coefficient (10−14~10−16 cm s−1) [10, 11]. To solve these problems, the most promising approach is carbon coating, which can enhance electronic conductivity, inhibit crystal growth, and provide channels to facilitate Li+ intercalation/deintercalation [12,13,14]. Furthermore, carbon source is one of the key factors of the carbon coating technology. The carbon layer on the particle surface has a significant influence on the electrochemical performance of LiFePO4, which is closely related to the electronic conductivity and structure of carbon [15,16,17,18,19,20]. Moreover, the uniform distribution of carbon facilitates the electrode reaction kinetics due to the full contact of active particles with each other [21,22,23,24,25]. Good electrical contact allows Li+ and electrons to be simultaneously obtained at the same position during charging/discharging process [26,27,28,29,30,31]. Therefore, the choice of carbon source is particularly significant.
In recent years, various organics have been used as carbon sources for LiFePO4/C composites, such as sucrose, glucose, and citric acid [32,33,34]. Among the various studies reported, the organics are decomposed into carbon under the inert gas protection of high temperature, which not only increases the electronic conductivity of LiFePO4, but also acts as a reductant [11, 35,36,37]. Moreover, various polymers have been reported as carbon sources of LiFePO4/C composites, such as polyvinyl alcohol (PVA) [38, 39], polystyrene (PS) [40], and polyethylene glycol (PEG) [37]. On one hand, H2 and carbon are produced by pyrolysis of polymers, which can be used as a reductant from Fe3+ to Fe2+. On the other hand, carbon coating on the surface of LiFePO4 increases its electronic conductivity and inhibits the growth of LiFePO4 crystallites [37,38,39,40]. Wang and co-workers have reported that PVA was used as carbon source for the carbothermal reduction of LiFePO4/C composites. It was found that the best electrochemical performance was achieved when the PVA content was 4% [38]. Nevertheless, the whole performance improvement is still confined, and the industrial applications still face severe challenges. Moreover, Zou and co-workers have reported that PVA gel has been used as carbon source in synthesis of LiFePO4/C composites [39]. The synthesis process was simple and commercially feasible in large-scaled production; however, the discharge capacities are still poor. Up to now, one of the most common synthesis methods of LiFePO4/C is carbothermal reduction with wet-mixing method. However, using this method should add lots of volatile organic compounds to obtain good mixing at the cost of serious pollution and high cost [41,42,43]. Although the simple operation and low cost using water as medium, there must be a drying process, thus resulting in bad mixing due to different density and solubility of various components [37]. After calcination, the synthesized of LiFePO4/C composites exhibit low electronic conductivity and poor electrochemical performance. Therefore, we need to find a way to solve these problems.
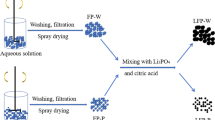
In this work, high-performance LiFePO4/C composites were synthesized by wet ball-milling, microwave drying, and carbothermal reduction with xylitol-PVA as complex carbon sources. The PVA is dissolved in water to form a hydrogel, and the precursors remain stable during the drying process without sedimentation and small deformation. Effects of the complex carbon sources on morphology, electronic conductivity, and electrochemical performance were analyzed by characterizations of X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscope (TEM), physical performance tests, and electrochemical techniques. High-performance LiFePO4/C cathode material has been realized with this method. At the same time, using xylitol as a monomer carbon source in the same synthesis as a control group.
Experimental
Materials synthesis
For comparison, the xylitol powders (AR, Beisitian, Tianjin) were fused as a monomer carbon source at 95 °C in an electronic multipurpose furnace. The fused xylitol, stoichiometric amounts of nano-scale FePO4 (AR, Qixing, Sichuan), and lithium carbonate (Li2CO3 AR, Zhongli, Sichuan) at molar ratio of 0.09:1:0.51 were ball-milled in a planetary mill with deionized water at 300 rpm for 1 h. The zirconia ball with a diameter of 0.8 mm was used for ball-milling and the ratio of ball to material was 6:1. The obtained slurry was subjected to microwave rapid drying (frequency, 2450 MHz), and then the precursor was transferred to a tube furnace at 750 °C for 4 h under the flowing nitrogen protection with a heating rate of 8 °C min−1 to synthesize LiFePO4/C composites, which was marked as LFP-1.
LiFePO4/C composites were synthesized through the method using the complex carbon sources as follows: Firstly, a solid content of 5 wt% PVA hydrogel was obtained by the PVA powders (Wanwei, Anhui, 1788) dissolved in deionized water at 90 °C in water bath. A slurry containing the precursors to the LiFePO4/C composites were synthesized by mixing the stoichiometric nano-scale FePO4, Li2CO3, and carbon sources at the same molar ratio in a planetary mill at 300 rpm for 1 h. Especially, the molar ratio of the fused xylitol and the PVA hydrogel is 3:1. For simplicity, the LiFePO4/C composites were synthesized by the same method, which was marked as LFP-2.
Material characterization
The crystalline structures were analyzed by XRD (D8 FOCUS Bruker AXS, Germany) with Cu Kα radiation in the 2θ scanning range of 10–80°. The surface morphology and microstructure were observed using SEM (FEI, Hong Kong) and TEM (JEOL, Japan). The carbon content was tested by the carbon sulfur tester (Eltra CS800). The particle size distribution was analyzed by the Laser Particle Sizer (LS609). The electronic conductivity was measured by two-point probe method. The specific surface area was measured using a B.E.T. apparatus (JW, Beijing, China).
Electrochemical measurements
The electrochemical measurements of the samples were measured using CR2032 coin-type half cells. The active material (LiFePO4/C), the conductive agent (acetylene black, AR, Xinlian, Henan), and the binder (polytetrafluoroethylene, AR, Chenguang, Sichuan) were uniformly mixed according to the weight ratio of 80:15:5 in absolute ethyl alcohol. After ultrasonic dispersion, a positive electrode film with a thickness of 0.14 mm was rolled. Finally, 10-mm diameter films were dried at 120 °C for 12 h under vacuum conditions. With a lithium foil as the counter and reference electrodes, CR2032 coin-type cells were assembled with a solvent mixture of 1 M LiPF6/ethyl carbonate (EC) + diethyl carbonate (DEC) (1:1, v/v) as the electrolyte (AR, Jinniu, Tianjin) under argon circulation protection in glove box. The galvanostatic charge/discharge tests were performed at the voltage range from 2.3 to 4.2 V (vs. Li/Li+) on a battery test system (LAND CT2001A, Wuhan, China) at 25 °C. Low-temperature electrochemical tests were performed in a high-low temperature box (Ronglixin Shenzhen, China) as follows: Firstly, after the initial two charge and discharge cycles at 0.2 C and 25 °C and fully charging, then the cells were placed in the set low-temperature environment (− 20 °C) for 15 h. Finally, the low-temperature discharge (cut-off voltage is 2.0 V) tests were at 0.2 C and 0.5 C. The cyclic voltammetry (CV) tests were performed at a scanning rate of 0.1 mV s−1 between 2.3 and 4.2 V on a CHI660C electrochemical workstation (Chenhua, Shanghai, China) at 25 °C. After two charge and discharge cycles at 0.2 C and 25 °C, the cells were charged to 50% DOC for electrochemical impedance spectroscopy (EIS) tests over the frequency range 0.1~1 × 105 Hz on the electrochemical workstation at 25 °C.
Results and discussion
Sample characterization
Under the sintering conditions of this study, both precursors have been successfully converted to an olivine structure, as shown in Fig. 1. Both samples exhibit the ideal orthorhombic olivine structure (JCPDS card number: 83-2092) without any impurity phase. The lattice parameters of the two samples are close to the standard parameters (a = 1.0329 nm, b = 0.6011 nm, c = 0.4699 nm, V = 0.29103 nm3). The strong and sharp peaks indicate a high crystallinity of the synthesized LiFePO4. Additionally, the carbon contents of the two samples are approximately 2.6 wt%, respectively, but no diffraction peak of carbon is found due to its existence in an amorphous form, which indicates that the presence of carbon has no effect on the crystal structure of LiFePO4. Furthermore, with the ratio of peak I111/I131 of the two samples changing, it is found that the ratio of LFP-2 sample increases to 0.8667 because of the complex carbon sources. The peak intensity ratio I111/I131 (R) can be used to measure the degree of cation mixing in the material. The higher the R value, the lower the degree of cation mixing in the material, which indicates good electrochemical performance of the material [44]. The R values of LFP-1 and LFP-2 samples are 0.8082 and 0.8667, respectively, implying the lower cation mixing degree of LFP-2 sample, which is beneficial to electrochemical performance.

XRD patterns of LiFePO4/C composites
The distribution of carbon has a significant effect on the morphology and microstructure of LiFePO4/C, as shown in Figs. 2 and 3. SEM observation shows that LFP-1 sample synthesized by xylitol as a monomer carbon source displays mostly irregular polygonal particles, and the average diameter of primary particle is 500 nm (Fig. 2a, b). The particle agglomeration may be prejudiced the electrochemical performance. However, LFP-2 sample obtained by xylitol and PVA as complex carbon sources exhibits spherical or spheroidal (Fig. 2c, d). The morphology of the particles is more regular without agglomeration. Moreover, the average size of primary particle is 350 nm, which is filled with a network-filamentous conductive carbon to enhance the electronic conductivity effectively among the particles. TEM observation shows that the LiFePO4 particles are surrounded by amorphous carbon, indicating that the carbon sources have been completely converted to amorphous carbon by pyrolysis (Fig. 3). LFP-1 sample is partly coated with carbon from xylitol, resulting in low electronic conductivity and excessive growth of crystal, as shown in Fig. 3a. However, LFP-2 sample is completely coated by carbon. Obviously, the amorphous carbon is uniformly distributed between LiFePO4 particles, which forms an effective conductive network to improve the diffusion of electrons and Li ions between the particles in Fig. 3b. Furthermore, the electronic conductivity was measured by a two-point probe method, which is 3.7 × 10−2 and 1.5 × 10−1 S cm−1 for LFP-1 and LFP-2, respectively. The carbon-coated LiFePO4/C demonstrates ~ 10−7–10−8 increase compared to pure LiFePO4 (~ 10−9 S cm−1). Furthermore, compared with LFP-1 sample, the electronic conductivity of LFP-2 sample demonstrates an order of magnitude improvement due to the PVA may provide more graphitic carbon [45]. The specific surface area was measured by the B. E. T. analysis, and LFP-2 sample exhibits the largest specific surface area of 19.738 m2 g−1 and increased by 17.3% compared with that of LFP-1 (16.329 m2 g−1). The higher specific surface area can be attributed to the smaller grain size. It is understandable that high specific surface area can facilitate its interfacial contact with the electrolyte, thus shortening the diffusion distance of Li+ and lowering the concentration polarization in the electrode [46]. The LiFePO4/C synthesized with the complex carbon sources features small grain size, high specific surface area, and superior electronic conductivity, which will thereby contribute to a superior electrochemical performance.

SEM images of LiFePO4/C composites a, b LFP-1 and c, d LFP-2

TEM images of LiFePO4/C composites a LFP-1 and b LFP-2
Electrochemical performances of LiFePO4/Li
To demonstrate the excellent electrochemical performance of the carbon-coated LiFePO4/C, the electrochemical tests of LFP-2 were compared with those of LFP-1. The initial charge/discharge curves of both samples at 0.2 C are shown in Fig. 4a. The first specific discharge capacity of LFP-2 sample reaches 162.2 mAh g−1 (the coulombic efficiency is 97.5%), while the specific discharge capacity of LFP-1 sample is 156.4 mAh g−1 (the coulombic efficiency is 94.5%). As shown in the inset of Fig. 4a, the polarization between charge/discharge platform at 0.5 Li insertion/extraction of LFP-1 and LFP-2 is 94.5 and 89.6 mV, respectively, indicating that the kinetics is optimal for the LiFePO4/C synthesized with the complex carbon sources [47]. Figure 4b shows the charge/discharge curves of both samples in the voltage range of 2.3–4.2 V at different rates of 0.2 C, 0.5 C, 1 C, 2 C, 5 C, and 10 C. As the discharge current density increases, the specific discharge capacity of the material decreases. Under various charge/discharge rates, LFP-2 sample shows significant higher discharge capacities than LFP-1 sample at various rates. As shown in Fig. 4b, the specific discharge capacity of LFP-1 sample decreased sharply from 156.4 mAh g−1 at 0.2 C to 109.1 mAh g−1 at 10 C, which may be attributed to its larger grain size and non-uniform carbon coating. However, LFP-2 sample shows superb rate capability with the specific discharge capacities of 162.2, 157.5, 152.9, 147.1, 134.5, and 119.7 mAh g−1 at 0.2 C, 0.5 C, 1 C, 2 C, 5 C, and 10 C, respectively. From 0.2 to 10 C, the discharge capacity loss of LFP-2 sample is only 26.2% (30.2% for LFP-1 sample), indicating that there is less polarization inside the electrode for the complex carbon sources sample, and the advantage becomes even apparent as the charge/discharge rates increases. The charge/discharge curves for both samples are similar, with flat platforms corresponding to the lithium extraction and insertion reactions, but they vary in platform potentials. As shown in Fig. 4c, the discharge platform potential of LFP-2 sample is 3.39, 3.37, 3.35, 3.31, 3.17, and 2.93 V at different current densities from 0.2 to 10 C, respectively, much higher than LFP-1 (3.38, 3.36, 3.34, 3.30, 3.15 and 2.91 V, respectively), indicating the lower polarization. These results can be attributed to the higher electronic conductivity, the smaller crystallite, and the higher specific surface area of the complex carbon sources.

a Charge/discharge curves at 0.2 C for the first cycle and amplified voltage flats (inset) at 0.2 C; b charge/discharge curves at different discharge rates; c dependence of middle discharge voltages; d rate capability of LiFePO4/C electrodes; e cycling performance curves at 5 C/5 C; f low-temperature performance at − 20 °C at 0.2 C and 0.5 C
Figure 4d compares the rate performances of the samples at different current densities. It can be seen that both samples exhibit rather reproducible capacities during 5-cycle test at each current rate. The specific capacity can be retrieved when the lower current rate (0.2 C) is applied again. This result confirms that the synthesized of LiFePO4/C exhibits stable. Under various charge/discharge rates, LFP-2 sample shows significant higher specific discharge capacities than LFP-1 sample at various rates. From 0.2 to 10 C, the specific discharge capacities of LFP-1 and LFP-2 samples are decreased by 30.2% and 25.6%, respectively. As the current rate turns back to 0.2 C, the specific discharge capacities quickly reach 155.8 and 160.6 mAh g−1 for LFP-1 and LFP-2 samples, respectively. The superior rate capability of LFP-2 sample can be ascribed to the improved electronic conductivity and the shortened ion diffusion distance in the composite. The result indicates that LFP-2 sample exhibits high electrochemical reversibility and structural stability, which are consistent with the CV measurement results (Fig. 5). Figure 4e demonstrates the cycle performance of LFP-1 and LFP-2 samples at 5 C and room temperature (25 °C). It can be seen that the initial specific discharge capacity of LFP-1 sample is 126.6 mAh g−1, and it still hosts the specific discharge capacity of 120.4 mAh g−1 (95.1% retention rate) after 500 cycles. Compared to LFP-1 sample, LFP-2 sample exhibits the higher initial specific discharge capacity (133.1 mAh g−1) and higher retention rate (approximately 100.0% retention rate) after 500 cycles, suggesting that the complex carbon sources can not only improve the specific discharge capacities at various rates, but also elevate the cycle performance of the materials.

CV curves of LiFePO4/C electrodes
The low-temperature performance has been one of the vital challenges for LiFePO4 to demonstrate high electrochemical performance in practical applications. It mainly depends on the conductivity of electrolyte at low temperature, the interface properties of electrode, and the diffusion ability of Li+ in the cathode material [48, 49]. Figure 4f demonstrates the discharge curves of both samples at 0.2 C and 0.5 C under low temperature of − 20 °C. As the discharge rate increases, the corresponding median potential at − 20 °C reduces dramatically. The discharge median potential of LFP-2 sample is approximately 3.3 V (3.2 V for LFP-1) at 0.2 C and 3.2 V (3.1 V for LFP-1) at 0.5 C. Obviously, the discharge median potential increases when the complex carbon sources were used. It can be seen that the specific discharge capacities of LFP-1 sample decreases from 105.6 to 73.7 mAh g−1, corresponding to 57.5% at 0.2 C and 48.6% at 0.5 C under 25 °C. In contrast, the specific discharge capacities of LFP-2 sample are 120.3 mAh g−1 at 0.2 C and 96.7 mAh g−1 at 0.5 C, corresponding to 74.2% at 0.2 C and 61.4% at 0.5 C under 25 °C, which exhibits better low-temperature performance. These results indicate that the synthesized LiFePO4/C by xylitol-PVA as complex carbon sources exhibits better low-temperature performance due to the higher electronic conductivity, high specific surface area, and smaller crystallite with the homogeneous carbon coating. Additionally, the conductive carbon filaments are closely connected between the particles, which shortens the diffusion distance of Li+ to enhance the mutual conversion between LiFePO4 and FePO4.
To investigate the property, mechanism, and electrode kinetic parameters of the electrode reaction, CV measurements were conducted at a scan rate of 0.1 mV s−1 over a voltage range of 2.3–4.2 V, as shown in Fig. 5. Each sample shows a set of peaks around 3.4 V, consisting of an oxidation peak (charge) and a reduction peak (discharge), which corresponds to Li+ intercalation/deintercalation. The CV profile of LFP-2 sample presents more symmetric and sharper redox-pair peaks than LFP-1 sample. Furthermore, LFP-2 sample exhibits a much lower redox peak potential difference φ1 (249 mV) than LFP-1 (329 mV), which indicates better reversibility. This suggests that LFP-2 sample exhibits significantly enhanced redox kinetics, which may be attributed to the regular fine particles, the uniform carbon coating, the reduced diffusion distance, and the improvement of electronic conductivity and high specific surface area. This indicates that LFP-2 sample exhibits excellent electrode kinetics and lower electrode polarization.
To analyze the diffusion dynamics, EIS tests of the half cells were carried out, and the corresponding Li+ diffusion coefficient was calculated by software fitting. The results are demonstrated in Fig. 6 and Table 1. Figure 6a shows the Nyquist plots for both samples and the inset is an equivalent circuit. Furthermore, the Nyquist plot is composed of a semicircle in high-frequency region and a straight line in low-frequency region. The intercept of the plot and the real axis Z’ represent the ohmic resistance (Re) of the cell, mainly attributed to the electrolyte and the module of the cell. Moreover, the semicircle in the high-frequency regions corresponds to the charge transfer impedance (Rct) due to the impedance caused by the migration of charge between the positive electrode material and the electrolyte interface. The straight line in the low-frequency region represents the Warburg impedance (W), which is caused by the diffusion of Li+ inside the LiFePO4/C cathode material [50, 51].

a Nyquist plots of both LiFePO4/C electrodes; b variation and fittings between Z’ and the reciprocal square root of the angular frequency in the low-frequency region
The plots fitting were performed based on the equivalent circuit using Z/view software. It can be seen from Fig. 6a that the charge transfer resistances of LFP-1 and LFP-2 samples are 40.8 and 20.2 Ω, respectively. It is clear that LFP-2 sample exhibits lower charge transfer resistance than LFP-1 sample, indicating that the complex carbon sources can reduce surface resistance and increase electronic conductivity. This clearly demonstrates that the homogeneous carbon coating and the regular fine particles not only form a highly efficient electron conduction network, but also effectively reduce SEI layer formation [52]. In addition, the lithium-ion diffusion coefficient is closely related to the Warburg coefficient (σ) is calculated according to the following formula:
where R represents the ideal gas constant, T represents the absolute temperature, A is the active area of the electrode, n is the number of electrons lost or reduced in each molecule, F is the Faraday constant, and C is the molar concentration of Li+ intercalated in the cathode material. Moreover, σ is the Warburg coefficient which is obtained by fitting a straight line composed of the low-frequency regions Z’ and ω−1/2. The slope of the fitted straight line is the Warburg coefficient σ of the discharge state, and ω is the angular frequency of the low-frequency region. The relationship between Z’ and ω−1/2 displayed under the formula [2].
Under the assumptions of negligible kinetic limitation associated with electrode resistances, the Warburg coefficient σ of LFP-1 and LFP-2 samples are 33.67 and 5.87 Ω s−1/2, respectively. Furthermore, Li+ diffusion coefficients of LFP-1 and LFP-2 samples can be calculated to be 3.35 × 10−13 and 1.21 × 10−11 cm2 s−1, respectively, as shown in Table 1. In contrast, LFP-2 sample shows two orders of magnitude higher than LFP-1 sample, which is attributed to regular fine particles and high specific surface area to shorten Li+ diffusion distance. Importantly, homogeneous carbon coating and carbon network among particles can enhance the electronic conductivity. Overall, these findings indicate that the synthesized LiFePO4/C exhibits excellent rate performance, cycle stability, and low-temperature performance.
Conclusions
In summary, the LiFePO4/C composite with uniform carbon coating was successfully synthesized by wet ball-milling, microwave drying, and carbothermal reduction using xylitol-PVA as complex carbon sources. It is found that the complex carbon sources yielded a pronounced effect on the morphology, microstructure, and the electrochemical performance of the LiFePO4/C. Specifically, the fused xylitol is readily coated on the surface of ferric phosphate, and the PVA hydrogel can maintain the precursors stable during the drying process, and the hydrogel also can be transformed into carbon coating around the LiFePO4 during calcination as the additional carbon source. The synthesized LiFePO4/C featured micron-sized particles, large specific surface area, and high electronic conductivity, which facilitated the ion/electron diffusion in the electrode and thereby yielded the excellent electrochemical performance. The synthesized LiFePO4/C exhibits high specific discharge capacities of 162.2 mAh g−1 at 0.2 C and 119.7 mAh g−1 at 10 C, as well as an excellent capacity retention approximately 100% at 5 C after 500 cycles. More strikingly, the specific discharge capacities at low temperature (− 20 °C) are 120.3 and 96.7 mAh g−1 at 0.2 C and 0.5 C, respectively. The approach of this study is simple and environmentally friendly, providing a broad prospect for large-scale commercial production.
References
Padhi AK, Nanjundaswamy KS, Goodenough JB (1997) Phospho-olivines as positive electrode materials for rechargeable lithium batteries. J Electrochem Soc 144:1188–1194
Wang Y, He P, Zhou H (2011) Olivine LiFePO4: development and future. Energy Environ Sci 4:805–817
Yuan LX, Wang ZH, Zhang WX, Hu XL, Chen JT, Huang YH, Goodenough JB (2011) Development and challenges of LiFePO4 cathode material for lithium-ion batteries. Energy Environ Sci 4:269–284
Wang J, Sun X (2015) Olivine LiFePO4: the remaining challenges for future energy storage. Energy Environ Sci 8:1110–1138
Zhang Y, Feng H, Wu X, Wang L, Zhang A, Xia T, Dong H, Liu M (2009) One-step microwave synthesis and characterization of carbon-modified nanocrystalline LiFePO4. Electrochim Acta 54:3206–3210
Jugovic D, Uskokovic D (2009) A review of recent developments in the synthesis procedures of lithium iron phosphate powders. J Power Sources 190:538–544
Yang Z, Dai Y, Wang S, Yu J (2016) How to make lithium iron phosphate better: a review exploring classical modification approaches in-depth and proposing future optimization methods. J Mater Chem A 4:18210–18222
Wang L, Liang G, Ou X, Zhi X, Zhang J, Cui J (2009) Effect of synthesis temperature on the properties of LiFePO4/C composites prepared by carbothermal reduction. J Power Sources 189:423–428
Castro L, Dedryvere R, El Khalifi M, Lippens PE, Breger J, Tessier C, Gonbeau D (2012) The spin-polarized electronic structure of LiFePO4 and FePO4 evidenced by in-lab XPS. J Phys Chem C 114:17995–18000
Ravet N, Chouinard Y, Magnan JF, Besner S, Gauthier M, Armand M (2001) Electroactivity of natural and synthetic triphylite. J Power Sources 97:503–507
Herle PS, Ellis B, Coombs N, Nazar L (2004) Nano-network electronic conduction in iron and nickel olivine phosphates. Nat Mater 3:147–152
Inagaki M (2012) Carbon coating for enhancing the functionalities of materials. Carbon 50:3247–3266
Oh SW, Myung ST, Oh SM, Oh KH, Amine K, Scrosati B, Sun YK (2010) Double carbon coating of LiFePO4 as high rate electrode for rechargeable lithium batteries. Adv Mater 22:4842–4845
Ding Y, Jiang Y, Xu F, Yin J, Ren H, Zhuo Q, Long Z, Zhang P (2010) Preparation of nano-structured LiFePO4/graphene composites by co-precipitation method. Electrochem Commun 12:10–13
Zhang Y, Park S-J (2018) Bimetallic AuPd alloy nanoparticles deposited on MoO3 nanowires for enhanced visible-light driven trichloroethylene degradation. J Catal 361:238–247
Zhang Y, Park S-J (2017) Au-pd bimetallic alloy nanoparticle-decorated BiPO4 nanorods for enhanced photocatalytic oxidation of trichloroethylene. J Catal 355:1–10
Zhang Y, Park S-J (2017) Incorporation of RuO2 into charcoal-derived carbon with controllable microporosity by CO2 activation for high-performance supercapacitor. Carbon 122:287–297
Zhang Y, Park M, Kim HY, Ding B, Park S-J (2016) In-situ synthesis of nanofibers with various ratios of BiOClx/BiOBry/BiOIz for effective trichloroethylene photocatalytic degradation. Appl Surf Sci 384:192–199
Zhang JH, Xie J, Wu CY, Cao GS, Zhao XB (2011) In-situ one-pot preparation of LiFePO4/carbon-nanofibers composites and their electrochemical performance. J Mater Sci Technol 27:1001–1005
Ma J, Li BH, Du HD, Xu CJ, Kang FY (2012) The improvement of the high-rate charge/discharge performances of LiFePO4 cathode material by Sn doping. J Solid State Electrochem 16:1353–1362
Nishimura S, Kobayashi G, Ohoyama K, Kanno R, Yashima M, Yamada A (2008) Experimental visualization of lithium diffusion in LixFePO4. Nat Mater 7:707–711
Tarascon JM, Armand M (2001) Issues and challenges facing rechargeable lithium batteries. Nature 414:359–367
Li H, Zhou H (2012) Enhancing the performances of Li-ion batteries by carbon-coating: present and future. Chem Commun 48:1201–1217
Yang GL, Jalbout AF, Xu Y, Yu HY, He XG, Xie HM, Wang RS (2008) Effect of polyacenic semiconductors on the performance of olivine LiFePO4. Electrochem Solid-State Lett 11:A125–A128
Wang M, Xue YH, Zhang KL, Zhang YX (2011) Synthesis of FePO4·2H2O nanoplates and their usage for fabricating superior high-rate performance LiFePO4. Electrochim Acta 56:4294–4298
Wang J, Sun X (2012) Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries. Energy Environ Sci 5:5163–5185
Yang KR, Lin ZJ, Hu XB, Deng ZH, Suo JS Preparation and electrochemical properties of a LiFePO4/C composite cathode material by a polymer-pyrolysis-reduction method. Electrochim Acta 56:2941–2946
Seid KA, Badot JC, Dubrunfaut O, Levasseur S, Guyomard D, Lestriez B (2012) Influence of the carboxymethyl cellulose binder on the multiscale electronic transport in carbon-LiFePO4 nanocomposites. J Mater Chem 22:24057–24066
Li BH, Xing YT, Chu XD, Ma J, He YB, Zhai DY, Du HD, Wei CG, Kang FY (2013) Effects of pyran-ring structure in carbon sources on the electrochemical performance of LiFePO4/C. Int J Electrochem Sci 8:446–457
Lin Y, Gao MX, Zhu D, Liu YF, Pan HG (2008) Effects of carbon coating and iron phosphides on the electrochemical properties of LiFePO4/C. J Power Sources 184:444–448
Zhang WJ (2011) Structure and performance of LiFePO4 cathode materials: a review. J Power Sources 196:2962–2970
Liang GC, Wang L, Ou XQ, Zhao X, Xu SZ (2008) Lithium iron phosphate with high-rate capability synthesized through hydrothermal reaction in glucose solution. J Power Sources 184:538–542
Wen L, Hu XD, Luo HZ, Li F, Cheng HM (2015) Open-pore LiFePO4/C microspheres with high volumetric energy density for lithium ion batteries. Particuology 22:24–29
Zhao Q, Zhang YZ, Meng Y, Wang YJ, Ou JK, Guo Y, Xiao D (2017) Phytic acid derived LiFePO4 beyond theoretical capacity as high-energy density cathode for lithium ion battery. Nano Energy 34:408–420
Hu LH, Wu FY, Lin CT, Khlobystov AN, Li LJ (2013) Graphene-modified LiFePO4 cathode for lithium ion battery beyond theoretical capacity. Nat Commun 4:1–7
Wu XL, Guo YG, Su J, Xiong JW, Zhang YL, Wan LJ (2013) Carbon-nanotube-decorated nano-LiFePO4/C cathode material with superior high-rate and low-temperature performances for lithium-ion batteries. Adv Energy Mater 3:1155–1160
Zhi XK, Liang GC, Ou XQ, Zhang SX, Wang L (2017) Synthesis and electrochemical performance of LiFePO4/C composite by improved solid-state method using a complex carbon source. J Electrochem Soc 164:A1285–A1290
Wang YQ, Liu ZP, Zhou SM (2011) An effective method for preparing uniform carbon coated nano-sized LiFePO4 particles. Electrochim Acta 58:359–363
Zou B, Wang Y, Zhou S (2013) Spray drying-assisted synthesis of LiFePO4/C composite microspheres with high performance for lithium-ion batteries. Mater Lett 92:300–303
Zhou Y, Gu CD, Zhou JP, Cheng LJ, Liu WL, Qiao YQ, Wang XL, Tu JP (2011) Effect of carbon coating on low temperature electrochemical performance of LiFePO4/C by using polystyrene sphere as carbon source. Electrochim Acta 56:5054–5059
Weng S, Yang Z, Wang Q, Zhang J, Zhang W (2013) A carbothermal reduction method for enhancing the electrochemical performance of LiFePO4/C composite cathode materials. Ionics 19:235–243
Ojczyk W, Marzec J, Swierczek K, Zajac W, Molenda M, Dziembaj R, Molenda J (2007) Studies of selected synthesis procedures of the conducting LiFePO4-based composite cathode materials for Li-ion batteries. J Power Sources 173:700–706
Kang H-C, Jun D-K, Jin B, Jin EM, Park K-H, Gu H-B, Kim K-W (2008) Optimized solid-state synthesis of LiFePO4 cathode materials using ball-milling. J Power Sources 179:340–346
Cho MY, Kim KB, Lee JW, Kim H, Kim H, Kang K, Roh KC (2013) Defect-free solvothermally assisted synthesis of microspherical mesoporous LiFePO4/C. RSC Adv 3:3421–3427
Doeff MM, Wilcox JD, Yu R, Aumentado A, Marcinek M, Kostecki R (2008) Impact of carbon structure and morphology on the electrochemical performance of LiFePO4 /C composites. J Solid State Electrochem 12:995–1001
Choi D, Kumta PN (2007) Surfactant based sol-gel approach to nanostructured LiFePO4 for high rate Li-ion batteries. J Power Sources 163:1064–1069
Saravanan K, Reddy MV, Balaya P, Gong H, Chowdari BVR, Vittal JJJ (2009) Storage performance of LiFePO4 nanoplates. Mater Chem 19:605–610
Liao XZ, Ma ZF, Gong Q, He YS, Pei L, Zeng LJ (2008) Low-temperature performance of LiFePO4/C cathode in a quaternary carbonate-based electrolyte. Electrochem Commun 10:691–694
Shin HC, Cho WI, Jang H (2006) Electrochemical properties of the carbon-coated LiFePO4 as a cathode material for lithium-ion secondary batteries. J Power Sources 159:1383–1388
Jin B, Jin EM, Park KH, Gu HB (2008) Electrochemical properties of LiFePO4-multiwalled carbon nanotubes composite cathode materials for lithium polymer battery. Electrochem Commun 10:1537–1540
Liu J, Jiang R, Wang X, Huang T, Yu A (2009) The defect chemistry of LiFePO4 prepared by hydrothermal method at different pH values. J Power Sources 194:536–540
Hwang J, Kong KC, Chang W, Jo E, Namd K, Kima J (2017) New liquid carbon dioxide based strategy for high energy/power density LiFePO4. Nano Energy 36:398–410
Funding
This work received financial support from the Natural Science Foundation of Hebei Province (Grant number E2015202356).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, J., Ren, X., Li, Z. et al. Synthesis and electrochemical performance of LiFePO4/C composite based on xylitol-polyvinyl alcohol complex carbon sources. Ionics 25, 1567–1575 (2019). https://doi.org/10.1007/s11581-018-2779-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-018-2779-9