
Abstract
Because enlarged exposed active planes facilitate the improvement of the rate capability of layered cathode material by providing more points for Li+ intercalation/de-intercalation, LiNi0.8Co0.15Al0.05O2 layered cathode material with enlarged exposed {010} active planes is desirable. In this article, micro-sized LiNi0.8Co0.15Al0.05O2 particles possessing enlarged exposed {010} active planes have been synthesized using Ni0.842Co0.158(OH)2 precursor prepared by an improved co-precipitation method. As-prepared LiNi0.8Co0.15Al0.05O2 demonstrates a good rate capability. The reversible capacity is 103 mAh g−1 at 10 °C, which is explained by the enhanced Li+ diffusion coefficient detected by CV and GITT measurements and reduced charge transfer resistance calculated by EIS tests. Moreover, the as-prepared LiNi0.8Co0.15Al0.05O2 also displays a good cycling stability. The capacity retention after 100 cycles at 0.2 °C is 85%. This good cycling stability can be ascribed to the porous structure of LiNi0.8Co0.15Al0.05O2 which provides a buffer area for volume expansion of the primary particles during cycling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Owing to their competitive cost, low toxicity, and high-energy density, Ni-rich layered cathode materials had drawn great attentions since ternary cathode materials were put forward by Ohzuku et al. in 2001 [1,2,3,4,5,6,7,8]. To date, some Ni-rich cathode materials such as LiNi0.8Co0.1Mn0.1O2 and LiNi0.8Co0.15Al0.05O2 have been successfully commercialized in consumer electronics industry and electric vehicles (EVs) one. Whereas, compared to LiCoO2 and Co-rich ternary cathode materials such as LiNi0.33Co0.33Mn0.33O2, Ni-rich layered cathode materials display much poor rate capabilities, which results in poor rate capabilities of LIBs [9, 10]. Resultantly, the poor rate capabilities of LIBs lead to a long charge time for EV. Therefore, in order to shorten the charge time of EV, a good strategy is to promote the rate capability of Ni-rich layered cathode materials since the rate capability of cathode materials has a great effect on the rate capabilities of LIBs.
For the sake of enhancing the rate capabilities of Ni-rich cathode materials, morphological designing, besides doping and surface modification, has been widely investigated because some particular morphologies are beneficial to enhancing the Li+ diffusion coefficient, leading to the improvement of rate capability of Ni-rich layered cathode materials [11,12,13,14,15,16,17,18,19,20,21]. For instance, nano-sized LiNi0.8Mn0.15Al0.05O2 particles have been synthesized and possess better capacity even at a high current density [22]. Nano-sized LiNi0.8Co0.1Mn0.1O2 particles have been fabricated and demonstrate an excellent rate capability [23]. With exception of reducing the particle size, enlarging exposed {010} active planes of Ni-rich layered cathode materials has also drawn great attentions because enlarging exposed {010} active planes perpendicular to Li+ diffusion tunnels can provide more points for intercalation/deintercalation of Li+ during cycling [24,25,26,27,28]. For example, spherical LiNi0.8Co0.15Mn0.05O2 particles possessing enlarged (010) active planes have been fabricated by a hydrothermal-calcination method [29]. LiNi0.7Co0.15Mn0.15O2 possessing enlarged exposed (010) active planes has been fabricated through a PVP auxiliary hydrothermal method using Ni0.7Co0.15Mn0.15(OH)2 as precursor [30]. Both Ni-rich layered cathode materials have a good rate capability. However, the as-prepared Ni-rich layered cathode materials cannot be put into practical use because of high cost of preparing method. Recently, Zhou et al. reported that Ni0.6Co0.2Mn0.2(OH)2 with thick primary plates prepared by a co-precipitation process facilitates the formation of the primary particles possessing enlarged exposed {010} planes for LiNi0.6Co0.2Mn0.2O2 [31]. The as-produced LiNi0.6Co0.2Mn0.2O2 demonstrates a good rate capability. Their results triggered us to try to prepare NCA possessing enlarged exposed {010} active planes by using Ni0.842Co0.158(OH)2 with thick primary plates. However, as shown in Fig. 2d, the Ni0.842Co0.158(OH)2 with thick primary plates cannot be fabricated by the conventional co-precipitation method.
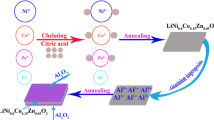
In this paper, we will display Ni0.842Co0.158(OH)2 with thick plates shown in Fig. 2b that can be synthesized by an improved co-precipitation method including two steps illustrated in Fig. 1. Moreover, NCA possessing enlarged exposed {010} active planes can be synthesized using Ni0.842Co0.158(OH)2 with thick primary plates [32]. And the as-prepared NCA demonstrates a highly improved rate capability and cycling stability.

A schematic diagram of preparing Ni0.842Co0.158(OH)2 with thick primary plates
Experimental
Synthesis of Ni0.842Co0.158(OH)2
Ni0.842Co0.158(OH)2 with thick plates was fabricated by an improved co-precipitation method. As illustrated in Fig. 1, the synthesis procedure is as follows. NiSO4·6H2O and CoSO4·7H2O were dissolved into a homogeneous solution at a stoichiometric ratio of Ni0.842Co0.158(OH)2 to form solution A (2 mol L−1). NaOH was dissolved in deionized water to form solution B (4 mol L−1). Solutions A, B, and ammonia water were slowly added into a reaction vessel to produce co-precipitates. At first, the reaction temperature was kept at around 30 °C. pH value was controlled at about 10.5. The concentration of ammonia in the reaction solution was kept at 1.5 mol L−1. After 2 h later, the reaction temperature was raised to 50 °C. The pH value was raised to 11.0. The concentration of ammonia in the reaction solution was decreased to 0.3 mol L−1. The reaction was lasted for 24 h. Afterwards, the co-precipitates were washed and dried. Ni0.842Co0.158(OH)2 particles synthesized under these reaction conditions were designated as T-precursor based on fact that the secondary Ni0.842Co0.158(OH)2 particle is consisted of the thick primary plates. Meanwhile, conventional spherical Ni0.842Co0.158(OH)2 particles were prepared under these reaction conditions of 50 °C temperature, pH value of 11.0, and the concentration of ammonia of 0.3 mol L−1. Ni0.842Co0.158(OH)2 particles synthesized under these reaction conditions were designated as S-precursor based on fact that the secondary Ni0.842Co0.158(OH)2 particle is spherical in shape.
Synthesis of NCA
NCA cathode materials were prepared as follows. Based on stoichiometric ratio of LiNi0.8Co0.15Al0.05O2, the dried precursor (T-precursor or S-precursor) was mixed with Al(NO3)3·9H2O and LiOH·H2O (excess of 2%). And then, the mixed powders were fired at 500 °C for 5 h and 765 °C for 16 h under O2 atmosphere to form LiNi0.8Co0.15Al0.05O2 which has an excellent crystalline structure. The samples synthesized using T-precursor and S-precursor as precursors were labeled as T-NCA and S-NCA, respectively.
Characterization
XRD spectra were achieved using X-ray diffractometer with Cu-Kα radiation to detect structures of precursor and cathode. The scanning scope was 10–80° at a speed of 0.02° s−1. SEM images were obtained using Hitachi S-4800 scanning electron microscopy to observe the morphologies and particle size of samples. Inductively, coupled plasma (ICP) measurements were performed to detect the compositions of samples. EDX images were gotten using energy-dispersive X-ray spectrometer to observe the elemental distribution. HR-TEM (high-resolution transmission electron microscopy) images were gained using FEI Tecnai G2 to verify the specific crystallographic planes. Mercury intrusion analysis using a mercury porosimetry was used to get pore size distribution of samples. XPS (X-ray photoelectron spectroscopy) spectra were achieved to detect the surface elemental oxidation state of samples.
Electrochemical measurement
The measurements for the electrochemical performances of prepared cathode were conducted using half cells. The details of preparing cells were present in previous literature [29]. The galvanostatic electrochemical data were obtained on LAND CT-2001A cell test instruments in a voltage range between 2.8 and 4.3 V at around 25 °C. Cyclic voltammograms (CV) and electrochemical impedance spectra (EIS) tests were conducted on an electrochemical workstation (Parstat 4000). Different voltage scan rates for the CV measurement are 0.05, 0.1, 0.2, and 0.25 mV s−1. The frequencies for the EIS measurement are from 100 kHz to 0.01 Hz and the fluctuation voltage is 10 mV.
Results and discussion
A comparison of morphology and structure between T-precursor and S-precursor was made to detect the differences between T-precursor and S-precursor. Figure 2 shows the SEM images and XRD patterns of T-precursor and S-precursor. Two precursors are obviously different in morphology. As shown in Fig. 2a–d, T-precursor demonstrates an irregular morphology for the secondary particles and thick plate-like one for the primary particles. However, S-precursor displays a spherical morphology for the secondary particles and thin plate-like one for the primary particles. As reported in previous literatures [27,28,29, 33], the thick plate-like morphology of the primary particle for T-precursor is speculated to be helpful to the formation of the primary particles possessing enlarged exposed (010) planes for T-NCA. Although they demonstrate different morphologies, two precursors present the similar XRD patterns. As displayed in Fig. 2e, all peaks can be indexed to the ophrastite structure of Ni(OH)2, indicating that two precursors have the same structure [30, 34].

SEM images (a–d) and XRD patterns (e) of T-precursor and S-precursor
The differences of morphology and structure between T-NCA and S-NCA were detected by SEM and XRD measurements. As shown in Fig. 3a–d, the morphologies of T-NCA and S-NCA particles are the same as the morphologies of T-precursor and S-precursor particles, respectively. The thick primary plates in the secondary particles for T-NCA can be clearly observed. In contrast, the thin primary plates in the secondary particles for S-NCA are present. Based on the conclusion in previous literatures [35, 36], in a thin plate shown in Fig. 3d, the faces with a large area (large faces) are supposed to be {003} inactive planes and those with a small area (small faces) are speculated to be {010} active planes. Therefore, in a thick plate shown in Fig. 3b, the area of large face decreases and one of small face increases, resulting in an increase of the area of exposed {010} active plane. This speculation will be proved by the observation through HR-TEM images (shown in Fig. 5). As shown in Fig. 3e, T-NCA and S-NCA demonstrate the similar XRD patterns [37]. The obvious separations of the (006)/(102) and (108)/(110) peaks manifest a well-crystallized structure, which is also supported by the high I(003)/I(104) value of 2.17 (T-NCA) and 2.11 (S-NCA) [38].

SEM images (a–d) and XRD patterns (e) of T-NCA and S-NCA
EDX-mapping measure measurements were conducted to identify the Ni, Co, and Al elements distribution. As shown in Fig. 4a–d, Ni, Co, and Al elements are homogeneously distributed in T-NCA particles, indicating Al element can be introduced into T-NCA particles by the high-temperature calcination. As shown in Fig. 4e–h, the Ni, Co, and Al elements are evenly distributed in S-NCA particles. Furthermore, XPS spectra were obtained to detect the surface elemental oxidation state of samples. As shown in Fig. S1a, b, and c, the binding energy peaks of Ni, Co, and Al for two samples are almost identical, indicating that the surface elemental oxidation states of both samples are same. As shown in Fig. S1, the Ni 2p3/2 peaks occur at 856.1 eV. The fitting Ni 2p3/2 spectrum exhibits two peaks at 856.9 and 855.4 eV which can be ascribed to Ni2+ and Ni3+, respectively. The Co 2p3/2 peaks occur at 780.1 eV, indicating the presence of Co3+. The Al 2p3/2 peaks occur at 73.8 eV, indicating the presence of Al3+. In addition, as listed in Table S1, the chemical compositions of T-NCA and S-NCA are in well agreement with those of LiNi0.8Co0.15Al0.05O2.

SEM (a) and EDX-mapping (b, c, d) images of T-NCA and SEM (e) EDX-mapping (f, g, h) images of S-NCA particles
TEM and HR-TEM measurements were conducted to further investigate the structure of T-NCA and S-NCA. As displayed in Fig. 5b, HR-TEM image was obtained from the edge of the primary particle (marked in a red circle) at a small secondary particle shown in Fig. 5a. HR-TEM image shows the clear sets of lattice fringes. The interplanar spacing of two lattice fringe is 4.72 Å, which corresponds to interplanar spacing of two (003) planes of layered cathode material. In previous literatures [39,40,41], the plane perpendicular to (003) plane was confirmed to be (010) plane. Therefore, it can be confirmed that T-NCA is consisted of the primary particles possessing some exposed {010} active planes parallel to small faces in the primary particles (As illustrated in Fig. 4b). As aforementioned, the ratio of area between small face and large one in a primary particle for T-NCA is larger than that for S-NCA. Accordingly, comparing with S-NCA, T-NCA has a larger area of face parallel to exposed {010} active planes. This deduction is further proved by HR-TEM measurement of S-NCA which hardly detects exposed {010} active planes for S-NCA. As displayed in Fig. S2c, HR-TEM image of S-NCA shows a closer lattice array compared to that of T-NCA. The interplanar spacing of two lattice fringe is 2.03 Å, which corresponds to interplanar spacing of two (003) planes of layered cathode material. It was expected that T-NCA has a better electrochemical properties than S-NCA since enlarged exposed {010} active planes are favorable to the intercalation/deintercalation of Li+ during cycling. In addition, a fast Fourier transform (FFT) image was also obtained. As showed in the lower right corner of Fig. 5c, an array of symmetry dots can be observed, indicating a single crystalline of primary particle. This result is in accord to that in previous literature [33].

TEM and HR-TEM images of T-NCA
To detect the electrochemical performances of T-NCA and S-NCA, the galvanostatic charge/discharge measurements were carried out under conditions of a voltage range between 2.8 to 4.3 V and 25 °C. The initial charge/discharge curves at 0.1 °C (1 °C = 190 mA g−1) are shown in Fig. 6a. The charge/discharge curve shapes for two samples are similar and in agreement with characteristics of Ni-rich materials. Nevertheless, the discharge capacity for T-NCA reaches to 195 mAh g−1 and the coloumbic efficiency is 87.3%. In contrast, the discharge capacity for S-NCA is 188 mAh g−1 and the coloumbic efficiency is less than 85%. Figure 6b shows the rate capabilities which were obtained by cycling cells at 0.1, 0.2, 0.5, 1.0, 3.0, 5.0, and 10.0 °C, gradually. Obviously, the rate capability of T-NCA is much better than that of S-NCA, which is in accord to above-mentioned speculation that the enlarged exposed {010} active planes of T-NCA are greatly helpful to improving rate capability. Figure 6c shows the cycling stabilities which were gotten by cycling cells at 0.2 °C for 100 cycles. As shown in Fig. 6c, the capacity retention of T-NCA after 100 cycles is 85% while that of S-NCA is 76%. As shown in Fig. 7a, b, the voltage decay of T-NCA after 100 cycles is also weaker than that of S-NCA, which is verified by dQ/dV curves shown in Fig. 7c, d. A main factor to the improved cycling stability for T-NCA can be explained as follows. As shown in SEM images in Fig. 3b, d, the pore size in the secondary particle for T-NCA is much larger than that for S-NCA, which is verified by pore size distribution curves shown in Fig. S3 and Table S2. As reported in previous literatures [42, 43], the micro-size pores in the secondary particle can provide a buffer area for volume expansion of the primary particles during cycling, which can eliminate the deterioration of cycling stability caused by the volume expansion.

Electrochemical properties of T-NCA and S-NCA: a initial charge-discharge curves at 0.1 °C, b rate capability, and c cycling stability at 0.2 °C

The charge-discharge curves of a T-NCA and b S-NCA at 0.2 °C at 1st, 50th, and 100th cycles. The dQ/dV curves of c T-NCA and d S-NCA
To further explore the origin of the improved electrochemical performances of T-NCA, CV, GITT, and EIS tests were conducted. CV measurements were operated in a voltage range between 2.8 and 4.3 V at scan different rates of 0.05, 0.10, 0.20, and 0.25 mV s−1, respectively. As shown in Fig. 8a, b, the peak currents increase when the scanning rates enhance. Moreover, as shown in Fig. 8c, d, the square root of the scanning rate and the peak current are linear correlation, suggesting that the diffusion coefficient of Li+ can be calculated using following equation [44, 45]:

Cyclic voltammograms at various voltage scan rates and chart of peak current ip of the cyclic voltammograms vs \( \sqrt{v} \): (a, c) T-NCA, (b, d) S-NCA
Where ipc represents the peak current, n represents the number of electrons per reaction species (one for Li+), F and R represent the Faraday constant and ideal gas constant, respectively. T and CLi+ are the experimental temperature and Li+ concentration in the cathode. DLi+ represents the diffusion coefficient of Li+. v and A represent the scan rate and surface area of electrode [46, 47]. The calculated values of diffusion coefficient of Li+ by the oxidation peak and reduction peak are listed in Table S3. DLi+ values for T-NCA electrode are higher than those for S-NCA electrode, which is in agreement with the highly improved rate capability of S-NCA. Besides, to investigate the dynamical processes of Li+ diffusion in the primary particles, GITT measurement is also conducted. GITT curves are shown in Fig. S4. And the calculated DLi+ values are listed in Table S4. DLi+ values of T-NCA are larger than those of S-NCA, which is in agreement with the improved rate capability of T-NCA. Therefore, improved rate capability of T-NCA can be mainly attributed to the enlarged exposed {010} planes. Moreover, EIS tests were also operated using cells after 100 cycles at 0.2 °C. Figure 9 shows the Nyquist plots of two samples. Two Nyquist plots demonstrate the similar shape, suggesting that the Nyquist plots can be fitted at the same equivalent circuit shown in Fig. 9. As reported in previous literature [48], Rs means the series resistance of electrolyte, RSEI is corresponded to the resistance of solid electrolyte interface film, and Rct means the charge transfer resistance between solid electrolyte interface film and active material. The fitted values of resistance are listed in Table S3, and values of RSEI and Rct for T-NCA are lower than those for S-NCA, which is in agreement with the enhanced cycling stability of T-NCA.

Nyquist plots of T-NCA and S-NCA electrodes after 100 cycles. Inset shows a corresponding equivalent circuit
Conclusion
In summary, LiNi0.8Co0.15Al0.05O2 possessing enlarged exposed {010} active planes can be synthesized using Ni0.842Co0.158(OH)2 precursor consisted of thick plates fabricated via an improved co-precipitation method. The as-prepared LiNi0.8Co0.15Al0.05O2 displays an outstanding rate capability. The discharge capacity reaches to 103 mAh g−1 at 10 °C. In addition, the improved co-precipitation method can be carried out by adjusting the process parameters in the process of co-precipitation reaction with no need of extra equipment. Therefore, this improved co-precipitation method is expected to be applied in large-scale production of cathode materials.
References
Liu W, Oh P, Liu X, Lee MJ, Cho W, Chae S, Kim Y, Cho J (2015) Nickel-rich layered lithium transition-metal oxide for high-energy lithium-ion batteries. Angew Chem 54:4440–4457
Hou P, Yin J, Ding M, Huang J, Xu X (2017) Surface/interfacial structure and chemistry of high-energy nickel-rich layered oxide cathodes: advances and perspectives. Small 4:1701802
Zheng JM, Wu XB, Yang Y (2011) A comparison of preparation method on the electrochemical performance of cathode material li[Li0.2Mn0.54Ni0.13Co0.13]O2 for lithium ion battery. Electrochim Acta 56:3071–3078
Yoon CS, Choi MH, Lim BB, Lee EJ, Sun YK (2015) Review—high-capacity Li[Ni1-xCox/2Mnx/2]O2 (x= 0.1, 0.05, 0) cathodes for next-generation li-ion battery. J Electrochem Soc 162:A2483–A2489
Robert R, Villevieille C, Novák P (2014) Enhancement of the high potential specific charge in layered electrode materials for lithium-ion batteries. J Mater Chem A 2:8589
Kim TH, Park JS, Chang SK, Choi S, Ryu JH, Song HK (2012) The current move of lithium ion batteries towards the next phase. Adv Energy Mater 2:860–872
Duan J, Wu C, Cao Y, Huang D, Du K, Peng Z, Hu G (2017) Enhanced compacting density and cycling performance of Ni-riched electrode via building mono dispersed micron scaled morphology. J Alloys Compd 695:91–99
Ohzuku T, Makimura Y (2001) Layered lithium insertion material of LiCo1/3Ni1/3Mn1/3O2 for lithium-ion batteries. Chem Lett 30:642–643
Manthiram A, Song B, Li W (2017) A perspective on nickel-rich layered oxide cathodes for lithium-ion batteries. Energy Storage Mater 6:125–139
Myung ST, Maglia F, Park KJ, Yoon CS, Lamp P, Kim SJ, Sun YK (2016) Nickel-rich layered cathode materials for automotive lithium-ion batteries: achievements and perspectives. ACS Energy Lett 2:196–223
Hua W, Wu Z, Chen M, Knapp M, Guo X, Indris S, Binder JR, Bramnik NN, Zhong B, Guo H, Chou S, Kang YM, Ehrenberg H (2017) Shape-controlled synthesis of hierarchically layered lithium transition-metal oxide cathode materials by shear exfoliation in continuous stirred-tank reactors. J Mater Chem A 5:25391–25400
Sheng T, Xu YF, Jiang YX, Huang L, Tian N, Zhou ZY, Broadwell I, Sun SG (2016) Structure design and performance tuning of nanomaterials for electrochemical energy conversion and storage. Acc Chem Res 49:2569–2577
Wang KX, Li XH, Chen JS (2015) Surface and interface engineering of electrode materials for lithium-ion batteries. Adv Mater 27:527–545
Zhou ZY, Tian N, Li JT, Broadwell I, Sun SG (2011) Nanomaterials of high surface energy with exceptional properties in catalysis and energy storage. Chem Soc Rev 40:4167–4185
Xie H, Du K, Hu G, Peng Z, Cao Y (2016) The role of sodium in LiNi0.8Co0.15Al0.05O2 cathode material and its electrochemical behaviors. J Phys Chem C 120:3235–3241
Chen M, Zhao E, Chen D, Wu M, Han S, Huang Q, Yang L, Xiao X, Hu Z (2017) Decreasing Li/Ni disorder and improving the electrochemical performances of Ni-rich LiNi0.8Co0.1Mn0.1O2 by Ca doping. Inorg Chem 56:8355–8362
Ju SH, Kang IS, Lee YS, Shin WK, Kim S, Shin K, Kim DW (2014) Improvement of the cycling performance of LiNi0.6Co0.2Mn0.2O2 cathode active materials by a dual-conductive polymer coating. ACS Appl Mater Interfaces 6:2546–2552
Ryu KS, Lee SH, Koo BK, Lee JW, Kim KM, Park YJ (2008) Effects of Co3(PO4)2 coatings on LiNi0.8Co0.16Al0.04O2 cathodes during application of high current. J Appl Electrochem 38:1385–1390
Liu Y, Lu Z, Deng C, Ding J, Xu Y, Lu X, Yang G (2017) A novel LiCoPO4-coated core–shell structure for spinel LiNi0.5Mn1.5O4 as a high-performance cathode material for lithium-ion batteries. J Mater Chem A 5:996–1004
Miao X, Ni H, Zhang H, Wang C, Fang J, Yang G (2014) Li2ZrO3-coated 0.4Li2MnO3·0.6LiNi1/3Co1/3Mn1/3O2 for high performance cathode material in lithium-ion battery. J Power Sources 264:147–154
Xu Y, Liu Y, Lu Z, Wang H, Sun D, Yang G (2016) The preparation and role of Li2ZrO3 surface coating LiNi0.5Co0.2Mn0.3O2 as cathode for lithium-ion batteries. Appl Surf Sci 361:150–156
Hwang I, Lee CW, Kim JC, Yoon S (2012) Particle size effect of Ni-rich cathode materials on lithium ion battery performance. Mater Res Bull 47:73–78
Li J, Yao R, Cao C (2014) LiNi1/3Co1/3Mn1/3O2 nanoplates with {010} active planes exposing prepared in polyol medium as a high-performance cathode for Li-ion battery. ACS Appl Mater Interfaces 6:5075–5082
Wei GZ, Lu X, Ke FS, Huang L, Li JT, Wang ZX, Zhou ZY, Sun SG (2010) Crystal habit-tuned nanoplate material of Li[Li1/3-2x/3NixMn2/3-x/3]O2 for high-rate performance lithium-ion batteries. Adv Mater 22:4364–4367
Zhao Y, Peng L, Liu B, Yu G (2014) Single-crystalline LiFePO4 nanosheets for high-rate Li-ion batteries. Nano Lett 14:2849–2853
Zhang L, Li N, Wu B, Xu H, Wang L, Yang XQ, Wu F (2015) Sphere-shaped hierarchical cathode with enhanced growth of nanocrystal planes for high-rate and cycling-stable li-ion batteries. Nano Lett 15:656–661
Zeng J, Cui Y, Qu D, Zhang Q, Wu J, Zhu X, Li Z, Zhang X (2016) Facile synthesis of platelike hierarchical Li1.2Mn0.54Ni0.13Co0.13O2 with exposed {010} planes for high-rate and long cycling-stable lithium ion batteries. ACS Appl Mater Interfaces 8:26082–26090
Yu R, Zhang X, Liu T, Xu X, Huang Y, Wang G, Wang X, Shu H, Yang X (2017) Hierarchically structured lithium-rich layered oxide with exposed active {010} planes as high-rate capability cathode for lithium-ion batteries. ACS Sustain Chem Eng 5:8970–8981
Wang Z, Liu H, Wu J, Lau WM, Mei J, Liu H, Liu GB (2016) Hierarchical LiNi0.8Co0.15Al0.05O2 plates with exposed {010} active planes as a high performance cathode material for Li-ion batteries. RSC Adv 6:32365–32369
Tian J, Su Y, Wu F, Xu S, Chen F, Chen R, Li Q, Li J, Sun F, Chen S (2016) High-rate and cycling-stable nickel-rich cathode materials with enhanced Li+ diffusion pathway. ACS Appl Mater Interfaces 8:582–587
Yang CK, Qi LY, Zuo Z, Wang RN, Ye M, Lu J, Zhou HH (2016) Insights into the inner structure of high-nickel agglomerate as high-performance lithium-ion cathodes. J Power Sources 331:487–494
Ruan Z, Zhu Y, Teng X (2015) Effect of pre-thermal treatment on the lithium storage performance of LiNi0.8Co0.15Al0.05O2. J Mater Sci 51:1400–1408
Wu N, Zhang Y, Guo Y, Liu S, Liu H, Wu H (2016) Flakelike LiCoO2 with exposed {010} facets as a stable cathode material for highly reversible lithium storage. ACS Appl Mater Interfaces 8:2723–2731
Makimura Y, Sasaki T, Nonaka T, Nishimura YF, Uyama T, Okuda C, Itou Y, Takeuchi Y (2016) Factors affecting cycling life of LiNi0.8Co0.15Al0.05O2 for lithium-ion batteries. J Mater Chem A 4:8350–8358
Fu F, Xu GL, Wang Q, Deng YP, Li X, Li JT, Huang L, Sun SG (2013) Synthesis of single crystalline hexagonal nanobricks of LiNi1/3Co1/3Mn1/3O2 with high percentage of exposed {010} active facets as high rate performance cathode material for lithium-ion battery. J Mater Chem A 1:3860
Xiao X, Yang L, Zhao H, Hu Z, Li Y (2011) Facile synthesis of LiCoO2 nanowires with high electrochemical performance. Nano Res 5:27–32
Yuan J, Wen J, Zhang J, Chen D, Zhang D (2017) Influence of calcination atmosphere on structure and electrochemical behavior of LiNi0.6Co0.2Mn0.2O2 cathode material for lithium-ion batteries. Electrochim Acta 230:116–122
Zhao J, Zhang W, Huq A, Misture ST, Zhang B, Guo S, Wu L, Zhu Y, Chen Z, Amine K, Pan F, Bai J, Wang F (2017) In situ probing and synthetic control of cationic ordering in Ni-rich layered oxide cathodes. Adv Energy Mater 7:1601266
Chen L, Su Y, Chen S, Li N, Bao L, Li W, Wang Z, Wang M, Wu F (2014) Hierarchical Li1.2Ni0.2Mn0.6O2 nanoplates with exposed {010} planes as high-performance cathode material for lithium-ion batteries. Adv Mater 26:6756–6760
Zhou YH, Wang Y, Li SM, Mei J, Liu H, Liu H, Liu GB (2017) Irregular micro-sized Li1.2Mn0.54Ni0.13Co0.13O2 particles as cathode material with a high volumetric capacity for Li-ion batteries. J Alloys Compd 695:2951–2958
Wu N, Wu H, Yuan W, Liu S, Liao J, Zhang Y (2015) Facile synthesis of one-dimensional LiNi0.8Co0.15Al0.05O2 microrods as advanced cathode materials for lithium ion batteries. J Mater Chem A 3:13648–13652
Kim J, Cho H, Jeong HY, Ma H, Lee J, Wang HJ, Park M, Cho J (2017) Self-induced concentration gradient in nickel-rich cathodes by sacrificial polymeric bead clusters for high-energy lithium-ion batteries. Adv Energy Mater 7:1602559
Zou Y, Yang X, Lv C, Liu T, Xia Y, Shang L, Waterhouse GI, Yang D, Zhang T (2017) Multishelled Ni-rich li(NixCoyMnz)O2 hollow fibers with low cation mixing as high-performance cathode materials for li-ion batteries. Adv Sci 4:1600262
Rho YH, Kanamura K (2003) Fabrication of thin film electrodes for all solid state rechargeable lithium batteries. J Electroanal Chem 559:69–75
Liu Y, Mi C, Yuan C, Zhang X (2009) Improvement of electrochemical and thermal stability of LiFePO4 cathode modified by CeO2. J Electroanal Chem 628:73–80
Hua N, Wang C, Kang X, Wumair T, Han Y (2010) Studies of V doping for the LiFePO4-based Li ion batteries. J Alloys Compd 503:204–208
Liu S, Yin H, Wang H, He J (2013) Electrochemical performance of WO2 modified LiFePO4/C cathode material for lithium-ion batteries. J Alloys Compd 561:129–134
Zhang Q, Peng T, Zhan D, Hu X (2014) Synthesis and electrochemical property of xLi2MnO3·(1−x)LiMnO2 composite cathode materials derived from partially reduced Li2MnO3. J Power Sources 250:40–49
Funding
The research was financially supported by the Sichuan Provincial Key Technology R&D Program (2016GZ0299).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(DOC 34789 kb)
Rights and permissions
About this article

Cite this article
Dong, H., Li, S., Liu, H. et al. Facile synthesis and electrochemical properties of LiNi0.8Co0.15Al0.05O2 with enlarged exposed active planes for Li-ion batteries. Ionics 25, 827–834 (2019). https://doi.org/10.1007/s11581-018-2620-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-018-2620-5