
Abstract
The RuxPd1-xO2 bimetallic electrocatalyst was synthesized by modifying Adams fusion method and used as the oxygen evolution reaction (OER) electrocatalyst in PEM water electrolysis. These synthesized RuxPd1-xO2 electrocatalysts morphology and electrochemical performances were characterized using FE-SEM, EDS, XRD, and cyclic voltammetry (CV) methods. The membrane electrode assemblies (MEAs) were fabricated using the synthesized RuxPd1-xO2 as the anode and 30% Pt/CB as a cathode and its electrochemical performance evaluated in single-cell PEM water electrolyzer at various experimental conditions and compared with pure RuO2. The results observed that the synthesized Ru0.8Pd0.2O2 electrocatalyst has shown better performance and stability compared to RuO2, a current density of 1 A/cm2 at the cell voltage of 2.03 V in 80 °C temperature. This synthesized Ru0.8Pd0.2O2 electrocatalyst can be used as the alternative to RuO2 at the oxygen evolution reaction (OER).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen is recognized as one of the most important components of the next-generation clean energy technology. Because of this, in recent years, hydrogen production and storage was attracting a lot of attention in both academia and industry due to its variety of applications in the energy sector. Within the whole cycle use of hydrogen energy, hydrogen production was considered as the key element of the upcoming hydrogen economy although there are several methods for the production of hydrogen [1,2,3,4]. The water electrolysis is the most sustainable method to produce hydrogen, but currently only 4% of hydrogen is produced by water electrolysis [5]. Nowadays, the proton exchange membrane water electrolysis (PEMWE) method has attracted significant importance for hydrogen production. PEM water electrolyzer was first introduced and developed in 1966 by General Electric Co., which was the most efficient method for hydrogen production from water and renewable energy sources, like wind or solar power at low temperature [2]. The produced hydrogen is a clean, zero carbon-free energy carrier, and the hydrogen economy comprises the production of hydrogen, storage, its transport, and finally the end use in fuel cells. Although PEM water electrolysis disadvantages include low efficiency and high cost of the material component such as membrane, bipolar plates, electrocatalyst, and higher anodic over potential for oxygen evolution reaction (OER) at typical operating current densities were the major limitation, in order to reduce the cost and to enhance the efficiency.
OER occurs on noble metal electrodes (Ir, Ru, Rh, Pt, Pd, Au) but, in general, metal oxides of IrO2 and RuO2 were found to be the most active electrocatalysts for oxygen evolution reaction compared to metal electrodes [6, 7]. RuO2 is a widely used material in electrochemical capacitors; also, it has a very high capacitance value of 150–260 mF/cm2 [6] as well as it is mostly used in the Chlor-alkali industry as a dimensionally stable anode (DSA). The high capacitance value of RuO2 arises from the pseudo-capacitance by the reaction of proton (H+) on the surface of RuO2 [8]. Mainly, the metal oxides described in the electrolysis process are based on the dimensionally stable anode (DSA) technology developed by H. Beer for the Chlor-alkali industry in 1965 [9]. The DSA-type electrodes of RuO2 and IrO2 were formed on titanium substrates by thermal decomposition of its precursors. However, ruthenium oxide (RuO2) is the most active electrocatalyst in OER, but its oxygen evolution activity is unfortunately not sufficient for long-term stability due to the increase in the oxidation state of ruthenium [10, 14].
Later combinations of RuO2 and IrO2 were studied as anode (OER) catalysts and found to have greater stability and activity; the Ir0.6Ru0.4O2 was shown to have the best performance by Marshall et al. [11]. But RuO2 and IrO2 are high-cost materials leading to the more expensive electrolyzer system. However, several non-noble metal oxides, such as TiO2, Ta2O5, and SnO2, were added to the IrO2 and RuO2 with increasing the stability and activity [8, 11,12,13]. In the last 15 years, higher performance for Nafion® PEM electrolyzers was reported, with the use of Ir, Ir-Ru, Pt-Ir, and Ir-Ta oxides, while Ir-Ru was shown to be the most active oxygen evolution electrocatalyst. Many ternary systems were also suggested as promising electrocatalysts for the oxygen evolution reaction [7, 15,16,17]. However, palladium (Pd) could be one such interesting substitute for anode electrocatalysts because of its relatively high electrocatalytic activity, stability, abundance, and low cost and is widely available on earth; although it has interesting electrocatalytic properties for various reduction and oxidation electrode processes [18,19,20,21], it has not been studied extensively for OER in PEM applications. RuO2 is a good electronic conductor, but a poor proton conductor, whereas palladium (Pd) is a proton conductor and has more conductivity in an anodic environment while adding palladium (Pd) to the RuO2 will act as network former, which increases the surface area of RuO2 and their activity for OER.
In the present study, RuxPd1-xO2 (x = 1, 0.8, 0.5, and 0.2) electrocatalysts were synthesized and studied for OER in PEM water electrolysis considering both electrochemical activity and stability. According to literature, no reports were found on RuxPd1-xO2, so we are attempting on this to observe the performance evaluation. The RuxPd1-xO2 materials were synthesized by modifying the Adams fusion method [15, 16, 22] and characterized by their structure, morphology, and electrochemical properties using X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM) with energy dispersive X-ray (EDS), and cyclic voltammetry.
Experimental methods
Materials
Palladium (II) chloride (PdCl2) and Ru (III) chloride (RuCl3.xH2O) purchased from Sigma Aldrich were used as precursors; NaNO3 (99.5% assay) and isopropyl alcohol (IPA) reagent grade purchased from Merck were used as reagent and solvent, respectively. Nafion® 115 membrane was procured from DuPont, USA.
Synthesis of electrocatalysts
The RuxPd1-xO2 (x = 1, 0.8, 0.5, and 0.2) electrocatalysts were synthesized by using a modified Adams fusion method [15, 16, 30]. The stoichiometric amount of chloride metal precursors of palladium chloride (PdCl2) and Ru (III) chloride (RuCl3·xH2O) was dissolved in IPA and stirred for 3 h [23, 24]. The total metal concentrations in the solution were approximately 0.05 M, while for the bimetallic oxides of Ru0.8Pd0.2O2, Ru0.5Pd0.5O2, and Ru0.2Pd0.8O2, to this solution, excess amount of NaNO3 was added and stirred well for 3 h. After that, the obtained solution was heated to 60 °C under continuous stirring until IPA evaporates. The resulted mixture was subjected to drying for 2 h in hot air oven at 80 °C and the dried sample was transferred into a silica crucible and calcinated in a muffle furnace at 500 °C for 1 h and cooled to room temperature. After cooling to room temperature, the resulting mixture was collected and washed with the excess amount of deionized water in order to remove all Cl− ions and dried at 80 °C in a vacuum oven overnight.
Membrane electrode assembly
The membrane electrode assembly (MEA) was fabricated by taking synthesized RuxPd1-xO2 as the anode catalyst and commercial 30 wt% Pt/CB (carbon black) as the cathode catalyst. Typically, the noble metal loadings on the membrane were maintained at 0.7 mg cm−2 for the cathode and 3.0 mg cm−2 for the anode [25]. The electrocatalyst ink solutions were prepared by mixing appropriate proportions of the electrocatalysts with 5 wt% Nafion® ionomer and IPA. The resulting mixture ink solution was sonicated for 60 min to make a homogenous dispersion. After that, the sonicated ink solution was sprayed on the Nafion® 115 membrane surface; one side is RuxPd1-xO2 and another side is Pt/CB, as well as Pt/CB sprayed on carbon cloth based on gas diffusion layer using a spraying gun and dried at room temperature to form a gas diffusion electrode (GDE). After completion of the coating, it was dried at room temperature for 30 min, and then hot pressed at 120 °C temperature, 60 kg cm−2 pressure for 3 min, to form a MEA; it was further tested in a PEM water electrolyzer [26].
PEM water electrolysis single cell operation
The fabricated MEA was placed in between two stainless steel (SS) plates with straight parallel flow fields to make an easy flow of reactants and products. The MEA of 25 cm2 active areas were assembled in their respective in-house fabricated PEMWE cell assemblies and were tightened with nuts and bolts with a torque 10 Nm2. The cell has a suitable inlet and outlet for circulation of DI water at the anode and cathode as well as for producing the gases hydrogen and oxygen at their particular electrodes. The PEMWE cell setup was provided with pencil heaters on both sides of the stainless steel end plates along with thermocouple and temperature controller for operating at different set temperatures. The DI water was circulated on both sides of the PEMWE cell using a pneumatic peristaltic pump at a flow rate of 60 ml min−1. A regulated DC power supply was used to supply power to the cell, and the voltage-current characteristics of the cell were manually measured as well as the hydrogen quantity.
Electrocatalyst characterization
The synthesized electrocatalysts morphology was carried out by using a field emission scanning electron microscope (FE-SEM). The elemental analysis of electrocatalysts was carried out by using a scanning electron microscope-energy-dispersive spectroscopy (SEM-EDS) model ZIESSLSM 510 Meta. XRD analysis of catalysts was carried out using PAN analytical X’Pert Pro MPD with Cu-Kα radiation between 2θ diffraction angles of 5–80° with a scan speed of 2 deg min−1.
The electrochemical studies were carried out using Gamry Reference 600 Potentiostat/Galvanostat in a three electrode cell assembly in the cyclic voltammetry (CV) method and linear sweep voltammetry (LSV) method, with a glassy carbon (GC) as working electrode (3 mm dia), saturated calomel electrode (SCE) and Pt wire as reference and counter electrode, respectively. All the experiments were carried out in 0.5 M H2SO4 solutions.
Results and discussion
Physicochemical characterizations of the electrocatalyst
The FE-SEM micrographs of Ru0.8Pd0.2O2 shows well crystalline particle structure and fine particles were shown in Fig. 1a, b. The Ru0.8Pd0.2O2 consists of fine particles with 50–80 nm, spherical in shape morphology with uniformity was observed. It appears that the particles were denser and agglomerated. Figure 1c, d shows the FE-SEM images of Ru0.5Pd0.5O2 and Ru0.2Pd0.8O2 nanoparticles were not uniform in shape and size.

a, b FE-SEM images of Ru0.8Pd0.2O2. c FE-SEM images of Ru0.5Pd0.5O2. d FE-SEM images of Ru0.2Pd0.8O2
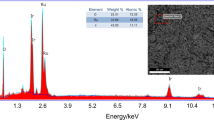
The SEM-EDS characterization of Ru0.8Pd0.2O2 were studied and represented in Fig. 2. From Fig. 2a, a well crystalline particle structure of Ru0.8Pd0.2O2 was observed.. However, SEM-EDS spectrum (Fig. 2b) of Ru0.8Pd0.2O2 confirmed the presence of ruthenium (Ru), palladium (Pd), and oxide (O) and elemental quantitative analysis of Ru0.8Pd0.2O2 was shown in Table 1.

a SEM-EDS images of Ru0.8Pd0.2O2. b EDS elemental analysis spectrum of Ru0.8Pd0.2O2
X-ray diffraction
The X-ray diffraction (XRD) spectral analysis of RuO2 and RuxPd1-xO2 were represented in Fig. 3. The peaks were indicated in the rutile structure of the electrocatalysts (JCPDS-880322) [18, 27,28,29]. The major diffraction peaks at 28, 35, 40, 54.3, 58.2, and 69.4o are the ruthenium oxide (RuO2) and was observed with (110), (101), (111), (110), (220) and (301), respectively. The peaks at 33.8, 40.01, and 68.03o started appearing upon addition of PdCl2; the peaks are identified to be palladium oxide (PdO) were observed with (111), (002), and (022), respectively [18, 25, 27, 28]. This complex formation was due to the reaction between PdCl2 and NaNO3 at a high temperature [23, 29]. The RuO2 peaks were clearly visible even in Pd-rich compositions, and it can be concluded that RuO2was well crystallizing in our experimental conditions. According to Terezo et al., Arikado et al., and Vinod kumar Puthiyapura et al., the differences in the crystal structure of RuO2 and PdO will make it difficult to form a solid solution. But in a mixed oxide system, in order to have an influence on catalyst activity, a perfect solid solution formation is not required, but a fine mixing of metals is sufficient [13, 29, 32].

XRD spectra of RuxPd1-xO2 electrocatalyst
The sharp, intense peaks confirm the crystalline phase of the synthesized RuO2 and RuxPd1-xO2 nanoparticles [30, 31]. All the X-ray diffraction peaks are corresponding to RuO2 and RuxPd1-xO2 only.
The average crystallite size of RuO2 and RuxPd1-xO2 was calculated according to the Debye–Scherrer’s equation, represented in Eq. (1):
where D indicates the average diameter of nm, k is the Scherrer constant (0.89), λ wavelength of X-rays (λ = 0.154 nm), is the full width at half maximum (FWHM) of the diffraction peaks and θ is the Bragg’s diffraction angle. Two major diffraction peaks of RuO2 and RuxPd1-xO2 at 28 and 54.3o were used to calculate the average crystalline size. The crystalline sizes of RuO2 and RuxPd1-xO2 were calculated from X-ray diffraction patrons and represented in Table 2. The highest crystalline size was found to be for RuO2; it is 5–6 nm; a gradual decrease in the crystalline sizes was evident on Pd addition. The crystalline size of RuO2 and Ru0.8Pd0.2O2 were almost similar due to the low Pd content. There was a steep decrease in crystalline size from Ru0.8Pd0.2O2 to Ru0.2Pd0.8O2 due to the higher content of Pd which has a very low ionic radius compared to Ru (IV). Lower crystalline sizes can indicate a higher geometrical surface area but do not essentially lead to electrochemical activity [12].
Electrochemical characterizations
Cyclic voltammetry
The cyclic voltammetry (CV) studies for RuO2 and RuxPd1-xO2 electrocatalysts were carried out using reference 600 potentiostat. Cyclic voltammetry studies of all prepared electrocatalysts were performed in 0.5 M H2SO4 solution with a scan rate of 10 m Vs−1at room temperature. The CV of RuO2 and RuxPd1-xO2 were represented in Fig. 4. The characteristic peaks of metal oxides at + 0.4 V and + 1.0 V (vs. SCE) is generally attributed to the Ru (III)/Ru(IV) and Ru(IV)/Ru(V) surface transitions, respectively, because of the redox charge transition between the RuO2 surface and hydrogen ion (H+) [27, 32,33,34,35].

CV of RuO2 and RuxPd1-xO2, reference electrode: SCE; working electrode: GC; counter electrode: Pt wire; scan rate, 10 mV s−1; electrolyte, 0.5 M H2SO4
The stability of the RuO2 and RuxPd1-xO2 electrocatalysts was tested using continuous CV studies within the potential range of + 0 to + 1.0 V (vs. SCE) which was represented in Fig. 5. From Fig. 5a, it was observed that after 100 cycles of potential scans, the capacitance, as well as the OER current, was decreased; this is due to the dissolution of RuO2 at the high anodic potential to form RuO4 which dissolves in the solution [36]. The characteristic peaks of RuO2 were decreased after several potential cycles and decrease in current density is gradual with cycles [29].

CV stability of the electrocatalysts a RuO2, b Ru0.8Pd0.2O2, c Ru0.5Pd0.5O2, and d Ru0.2Pd0.8O2
The CV of the Ru0.8Pd0.2O2 oxide was represented in Fig. 5b, where the corresponding peaks of Ru redox couples appear. The Ru0.8Pd0.2O2 catalyst shows the higher stability compared with RuO2 during 100 cycles due to the addition of Pd was found to stabilize the RuO2 in the modified Adams fusion method; this method gives better mixture formation of the bimetallic system [29]. Also, the decreased oxygen evolution current was lower by the addition of Pd compared to pure RuO2.
Figure 5c shows the CV of the Ru0.5Pd0.5O2 electrocatalyst; this catalyst exhibits lower current density and stability for the oxygen evolution reaction during repetitive100 cycles. The oxygen evolution reaction current density was gradually decreased after several potential cycles denotes a degradation of the structure which might be related to electrocatalyst dissolution [23].
The characteristic CV for the Ru0.2Pd0.8O2 electrocatalyst was represented in Fig. 5d. This catalyst contains the highest amount of Pd (80% atomic ratio) and reveals the lowest performance for the oxygen evolution reaction and less stability during repetitive 100 cycles. [32] The characteristic peaks of RuO2 were lost after several potential cycles, and decrease in current density are gradual with cycles, which clearly indicates the dissolution of the RuO2 [36, 37].
Linear sweep voltammetry
The linear sweep voltammetry (LSV) studies were preformatted in 0.5 M H2SO4 with a scan rate of 10 mV s−1, in the interval 1.1–1.5 V (SCE) corresponding to the oxygen evolution reaction (OER). LSV curves for the entire series of the RuxPd1-xO2 electrocatalysts were represented in Fig. 6. From Fig. 6, the pure RuO2 is the most active for the OER, among the onset potential for OER at 1.15 V. By increasing the Pd content in the mixed oxides, the electrocatalytic activity in terms of current density was decreased, even though the onset potential for the OER remains the same for Ru0.8Pd0.2O2 and Ru0.5Pd0.5O2 electrocatalysts. The Ru0.2Pd0.8O2 electrocatalyst exhibits the lowest performance in the oxygen evolution region.

Linear sweep polarization curves of RuxPd1-xO2 electro catalysts, with scan rate 10 mV s−1 in the interval of 1.1–1.5 V (vs. SCE) in 0.5 MH2SO4
Performance of RuxPd1-xO2 in PEM water electrolysis
The synthesized ruthenium palladium oxide (RuxPd1-xO2) was used as oxygen evaluation electrocatalyst at the anode and 30 wt% Pt/C as a cathode for the fabrication of membrane electrode assembly (MEA). The fabricated MEA’s performance was tested in PEM water electrolysis single cell assemblies in 25 cm2 active areas. The electrochemical performance and characteristics of the prepared electrocatalysts were evaluated using distilled water along with corresponding yields of hydrogen production (Table 3) at different temperatures, 30, 40, 50, 60, 70, and 80 °C and at different current densities from 0.1 to 2.0 A/cm2 were studied and represented in Fig. 7. It shows that the current density and hydrogen production rate was increased with increasing the cell temperatures at all cell voltages. It might be recognized to the improved electrocatalytic activity and decreased in cell ohmic resistance [38]. The faradaic efficiency of the prepared RuxPd1-xO2 electrocatalysts for the oxygen evolution reaction(OER) has been calculated using the experimental hydrogen yield and theoretical hydrogen yield(assuming 100%). The calculated faradaic efficiency was obtained approximately 85–90% as shown in Table 3 because of it maybe cell contact resistance, internal current losses, gas measuring errors, and gap between water and gas in the gas liquid separators.

Current-voltage polarization of Ru0.8Pd0.2O2 in 25 cm2 single cell assembly at various temperatures 30, 40, 50, 60, 70, and 80 °C
The performance of the synthesized RuxPd1-xO2 catalyzed MEAs (Ru0.8Pd0.2O2, Ru0.5Pd0.5O2, and Ru0.2Pd0.8O2) were compared with pure RuO2 catalyzed MEAs in PEM water electrolysis cell at 80 °C with different current densities as shown in Fig. 8. However, the Ru0.8Pd0.2O2 electrocatalyst was shown with better electrochemical performance while compared to pure RuO2. It might be due to the better structural, morphological, surface characteristics of the electrocatalyst and possibly to the higher catalytic activity of the substrate. The cell voltages obtained with the RuO2, Ru0.8Pd0.2O2, Ru0.5Pd0.5O2, and Ru0.2Pd0.8O2 were observed to be 2.04, 2.03, 2.15, and 2.22 V respectively, at the operating current density of 1000 mA/cm2 (1 A/cm2). The obtained results show that the synthesized Ru0.8Pd0.2O2 electrocatalyst could be considered as an alternative to RuO2 toward the oxygen evolution reaction (OER) in PEM water electrolysis. The efficiency of the Ru0.8Pd0.2O2 catalyzed MEA in PEM water electrolyser was found to be 74% at a constant current density of 1 A/cm2 at 80 °C temperature. The efficiency was calculated by Gibbs free energy equation [39].

Performance of different MEA’s in 25 cm2 PEMWE cell at 80 °C
Further, the stability studies of PEM water electrolysis cell were carried out with Ru0.8Pd0.2O2 catalyzed MEA at a constant current density of 1 A/cm2 at 80 °C temperature; the results are shown in Fig. 9. The observed cell voltage of 2.03 V was almost stable during 100 h of continuous operation; it reveals the higher stability of Ru0.8Pd0.2O2 catalyzed MEA for the oxygen evolution reaction (OER) in PEM water electrolysis. From the qualitative point of view, the obtained results using Ru0.8Pd0.2O2 for the OER at the anode exhibited better efficiency compared to those obtained with RuO2.

Performance of Ru0.8Pd0.2O2 MEA in 25 cm2 single cell in constant current of 1 A cm−2 at 80 °C
Conclusion
In the present study, bimetallic RuxPd1-xO2 electrocatalysts were synthesized by modifying the Adams fusion method and used as the OER electrocatalyst at the anode in PEM water electrolyzer for hydrogen production. The synthesized Ru0.8Pd0.2O2 electrocatalyst showed similar crystallographic properties, morphology, and particle size of the RuO2 catalyst. The obtained results revealed that the synthesized Ru0.8Pd0.2O2 electrocatalyst has shown better electrochemical activity and stability compared to RuO2. It indicates that the Ru0.8Pd0.2O2 can be used as an alternative to RuO2 for the OER in PEM water electrolyzer. The present study could potentially be used in other PEM cells such as unitized regenerative fuel cells and hydrogen fuel cells.
References
Nikolaidis P, Poullikkas A (2017) A comparative overview of hydrogen production processes. Ren and Sus Energy Reviews 67:597–611. https://doi.org/10.1016/j.rser.2016.09.044
Ma L, Sui S, Zhai Y (2009) Investigations on high performance proton exchange membrane water electrolyzer. Int J Hydrogen Energy 34:678–684 doi.org/10.1016/j.ijhydene.2008.11.022
Momirlan M, Veziroglu TN (2002) Current status of hydrogen energy. Ren and Sus Energy Reviews 6(1-2):141–179. https://doi.org/10.1016/S1364-0321(02)00004-7
Shiva Kumar S, Ramakrishna SUB, Srinivasulu Reddy D, Bhagawan D, Himabindu V (2017) Synthesis of polysulfone and zirconium oxide coated asbestos composite separators for alkaline water electrolysis. Chem Eng Process Tech 3(1):1035
Dunn S (2002) Hydrogen futures: toward a sustainable energy system. Int J Hydrog Energy 27(3):235–264. https://doi.org/10.1016/S0360-3199(01)00131-8
Santana MHP, De Faria LA (2006) Oxygen and chlorine evolution on RuO2 + TiO2 + CeO2+ Nb2O5 mixed oxide electrodes. Electrochim Acta 51:3578–3585. https://doi.org/10.1016/j.electacta.2005.09.050
Baglio V, Di Blasi A, Denaro T, Antonucci V, Arico AS, Ornelas R et al (2008) Synthesis, characterization and evaluation of IrO2-RuO2 electrocatalytic powders for oxygen evolution reaction. J New Mat Electr Sys 11:105–108
Kotz R, Stucki S (1986) Stabilization of RuO2 by IrO2 for anodic oxygen evolution in acid media. Electrochim Acta 31:1311–1316. https://doi.org/10.1016/0013-4686(86)80153-0
Beer HB (1980) The invention and industrial development of metal anodes. J of the Electrochem Society 127(8):303C–307C. https://doi.org/10.1149/1.2130021
Sedlak JM, Lawrance RJ, Enos JF (1981) Advances in oxygen evolution catalysis in solid polymer electrolyte water electrolysis. Int J Hydrogen Energy 6:159–165. https://doi.org/10.1016/0360-3199(81)90004-5
Marshall AT, Sunde S, Tsypkin M, Tunold R (2007) Performance of a PEM water electrolysis cell using IrxRuyTazO2 electrocatalysts for the oxygen evolution electrode. Int J Hydrogen Energy 32:2320–2324. https://doi.org/10.1016/j.ijhydene.2007.02.013
Wu X, Tayal J, Basu S, Scott K (2011) Nano-crystalline RuxSn1-xO2 powder catalysts for oxygen evolution reaction in proton exchange membrane water electrolysers. Int J Hydrogen Energy 36:14796–14804. https://doi.org/10.1016/j.ijhydene.2011.01.067
Terezo AJ, Pereira EC (1999) Preparation and characterization of Ti/RuO2-Nb2O5 electrodes obtained by polymeric precursor method. Electrochim Acta 44:4507–4513. https://doi.org/10.1016/S0013-4686(99)00182-6
Galizzioli D, Tantardini F, Trasatti S (1974) Ruthenium dioxide: a new electrode material. I. Behaviour in acid solutions of inert electrolytes. J Appl Electrochem 4(1):57–67. https://doi.org/10.1007/BF00615906
Cheng J, Zhang H, Chen G, Zhang Y (2009) Study of IrxRu1-xO2 oxides as anodic electrocatalysts for solid polymer electrolyte waterelectrolysis. Electrochim Acta 54:6250–6256. https://doi.org/10.1016/j.electacta.2009.05.090
Adams R, Shriner RL (1923) Platinum oxide as a catalyst in the reduction of organic compounds. III. Preparation and properties of the oxide of platinum obtained by the fusion of chloroplatinic acid with sodium nitrate. J Am Chem Soc 45(9):2171–2179. https://doi.org/10.1021/ja01662a022.
Chanda D, Hnat J, Bystron T, Paidar M, Bouzek K (2017) Optimization of synthesis of the nickel-cobalt oxide based anode electrocatalyst and of the related membrane-electrode assembly for alkaline water electrolysis. Journal of Power Sources 347:247–258. https://doi.org/10.1016/j.jpowsour.2017.02.057
Baylet A, Marecot P, Duprez D, Castellazzi P, Groppi G, Forzatti P (2011) In situ Raman and in situ XRD analysis of PdO reduction and Pdo oxidation supported on γ-Al2O3 catalyst under different atmospheres. Phys Chem Chem Phys 13(10):4607–4613. https://doi.org/10.1039/c0cp01331e
Grigoriev SA, Millet P, Fateev VN (2008) Evaluation of carbon supported Pt and Pd nanoparticles for the hydrogen evolution reaction in PEM water electrolysers. J Power Sources 177:281–285. https://doi.org/10.1016/j.jpowsour.2007.11.072
Grigoriev SA, Lyutikova E, Martemianov S, Fateev VN, Lebouin C, Millet P (2006) Palladium-based electrocatalysts for PEM applications. WHEC Lyon Fr 16:13–16
Lu Y, Jiang Y, Gao X, Wang X, Che W (2014) Strongly coupled Pd nanotetrahedron/tungsten oxide nanosheet hybrids with enhanced catalytic activity and stability as oxygen reduction electrocatalysts. J Am Chem Soc 136:11687–11697. https://doi.org/10.1021/ja5041094
Song SD, Zhang HM, Ma XP, Shao ZG, Zhang YN, Yi BL (2006) Bifunctional oxygen electrode with corrosion-resistive gas diffusion layer for unitized regenerative fuel cell. Electro chem. Commun 8:399–405. https://doi.org/10.1016/j.elecom.2006.01.001
Papazisi KM, Siokou A, Balomenou S, Tsiplakides D (2013) Preparation and characterization of IrxPt1-xO2 anode electrocatalysts for the oxygen evolution reaction. Int J Hydrogen Energy 37:16642–16648. https://doi.org/10.1016/j.ijhydene.2012.02.118
Audichona T, Guenota B, Barantona S, Cretinb M, Lamyb C, Coutanceaua C (2017) Preparation and characterization of supported RuxIr(1-x)O2 nano-oxides using a modified polyol synthesis assisted by microwave activation for energy storage applications. Appl Catalysis B: Environ 200:493–502. https://doi.org/10.1016/j.apcatb.2016.07.048
Ramakrishna SUB, Srinivasulu Reddy D, Shiva Kumar S, Himabindu V (2016) Nitrogen doped CNTs supported palladium electrocatalyst for hydrogen evolution reaction in PEM water electrolyser. Int J Hydrogen Energy 41:20447–20454. https://doi.org/10.1016/j.ijhydene.2016.08.195
Sasikumar G, Muthumeenal A, Pethaiah SS, Nachiappan N, Balaji R (2008) Aqueous methanol electrolysis using proton conducting membrane for hydrogen production. Int J Hydrogen Energy 33:5905–5910. https://doi.org/10.1016/j.ijhydene.2008.07.013
Naga Mahesh K, Balaji R, Dhathathreyan KS (2016) Palladium nanoparticles as hydrogen evolution reaction (HER) electrocatalyst in electrochemical methanol reformer. Int J Hydrogen Energy 41:46–51. https://doi.org/10.1016/j.ijhydene.2015.09.110
Beck G, Bachmann C, Bretzler R, Kmeth R (2014) Thermal stability of platinum, palladium and silver films on yttrium-stabilised zirconia. Thin Solid Films 573:164–175. https://doi.org/10.1016/j.tsf.2014.11.035
Puthiyapura VK, Pasupathi S, Basu S, Wua X, Su H, Varagunapandiyan N, Pollet B, Scott K (2013) RuxNb1-xO2 catalyst for the oxygen evolution reaction in proton exchange membrane water electrolysers. Int J Hydrogen Energy 38:8605–8866. https://doi.org/10.1016/j.ijhydene.2013.04.100
Khorasani-Motlagh M, Noroozifar M, Yousefi M (2011) A simple new method to synthesize Nano crystalline ruthenium dioxide in the presence of octanoic acid as organic surfactant. Int J Nanosci Nanotechnol 7:167–172
Audichon T, Mayousse E, Napporn TW, Morais C, Comminges C, Boniface Kokoh K (2014) Elaboration and characterization of ruthenium nano-oxides for the oxygen evolution reaction in a proton exchange membrane water electrolyzer supplied by a solar profile. Electrochim Acta 132:284–291. https://doi.org/10.1016/j.electacta.2014.03.141
Arikado T, Iwakura C, Tamura H (1977) Electrochemical behavior of the ruthenium oxide electrode prepared by the thermal decomposition method. Electrochim Acta 22:513–518. https://doi.org/10.1016/0013-4686(77)85114-1
Zheng JP, Cygan PJ, Jow TR (1995) Hydrous ruthenium oxide as an electrode material for electrochemical capacitors. J Electrochem Soc 142(8):2699–2703. https://doi.org/10.1149/1.2050077
Trasatti S, Buzzanca G (1971) Ruthenium dioxide: a new interesting electrode material. Solid state structure and electrochemical behaviour. J Electroanal Chem Interfacial Electrochem 29:A1–A5. https://doi.org/10.1016/S0022-0728(71)80111-0
Ardizzone S, Fregonara G, Trasatti S (1990) “Inner” and “outer” active surface of RuO2 electrodes. Electrochimica Acta 35:263–267. https://doi.org/10.1016/0013-4686(90)85068-X
Song S, Zhang H, Ma X, Shao Z, Baker RT, Yi B (2008) Electrochemical investigation of electrocatalysts for the oxygen evolution reaction in PEM water electrolyzers. Int J Hydrogen Energy 33:4955–4961. https://doi.org/10.1016/j.ijhydene.2008.06.039
Grigoriev SA, Millet P, Dzhus KA, Middleton H, Saetre TO, Fateev V (2010) Design and characterization of bi-functional electrocatalytic layers for application in PEM unitized regenerative fuel cells. Int J Hydrogen Energ 35:5070–5076. https://doi.org/10.1016/j.ijhydene.2009.08.081
Abdin Z, Webb CJ, Gray E, Mac A (2015) Modelling and simulation of a proton exchange membrane (PEM) electrolyser cell. Int J Hydrogen Energy 40:13243–13257. https://doi.org/10.1016/j.ijhydene.2015.07.129
Faruk SO, Fatih B, Mat Mahmut D, Yuksel K (2011) Development and testing of a highly efficient proton exchange membrane (PEM) electrolyzer stack. Int J Hydrogen Energy 36:11480–11487. https://doi.org/10.1016/j.ijhydene.2011.01.129
Acknowledgements
The authors would like to express their sincere thanks to the Bhabha Atomic Research Centre (BARC), Board of Research in Nuclear Sciences (BRNS), Department of Atomic Energy (DAE), Government of India, for providing financial support for this work (Project Sanction Order No: 2013/36/21/BRNS/1739).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shiva Kumar, S., Ramakrishna, S.U.B., Bhagawan, D. et al. Preparation of RuxPd1-xO2 electrocatalysts for the oxygen evolution reaction (OER) in PEM water electrolysis. Ionics 24, 2411–2419 (2018). https://doi.org/10.1007/s11581-017-2359-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-017-2359-4