
Abstract
Misfit-layered calcium cobaltites (Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9), as anode materials for lithium-ion batteries, were synthesized by a simple hydro-decomposition method. All synthesized samples do not show any impurity phase. They exhibited plate-like particle with the particle size of 1–2 μm. The specific capacities of doped samples showed higher electrochemical performance compared to the undoped sample. After charge/discharge of 50 cycles, the specific capacities of Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 were 343, 562, and 581 mAh g−1, respectively. The doped samples showed an increase of over 60% compared to the undoped sample. The cyclic voltammetry profile of the doped samples showed the enhanced reactivity corresponding to their improved electrochemical performance. The capacity improvement of doped samples resulted from the metal oxide/Li conversion reactions, volume change, and high reactivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rechargeable lithium-ion batteries (LIBs) are extensively used as energy storage device for modern electronic devices. The LixCoO2 is the commercially cathode materials while graphite is widely used as anode material in LIBs [1, 2]. However, they were limited by the theoretical capacity of 140 and 372 mAh g−1, respectively [1, 2]. Hence, the alternative materials have been considered and researched. Especially, the metal oxide materials have been attractive such as LiMn2O4, V2O5 Fe2O3, Co3O4, TiO2, MnO for replacing LixCoO2 and graphite owing to their high theoretical capacity [1, 3–15].
Misfit-layered calcium cobalt oxide (Ca3Co4O9) has been attractively investigated over the years because of its interesting physical and chemical properties [16–19]. Several studies have been researched on its crystal information [16, 20], electronic structure [21], and magnetic properties [16, 22]. In fact, Ca3Co4O9 is better expressed as [Ca2CoO3][CoO2]1.61 since the crystal structure consists of two monoclinic sublattices: a Ca2CoO3 rock-salt layer and a CdI2-type CoO2 layer, stacking alternatively along the c-axis [16]. In addition, a large thermoelectric power of 125 μV/K at 300 K was reported in a single crystal Ca3Co4O9 [16]. This leads to intensive research on thermoelectric properties of this material [16–19]. Recently, Ca3Co4O9 was reported to show the giant dielectric property with a high dielectric permittivity of 1.38 × 104 at 20 °C and 1 kHz [23].
The electrochemical property of the Ca3Co4O9/Li half-cell was firstly reported by Kim et al. [24], with a large discharge capacity of 500 mAh g−1 after 50 cycles, due to its high theoretical capacity of 643.3 mAh g−1. The Ca3Co4O9 anode was decomposed into Co nanoparticles dispersed in the Li2O-CaO matrix after the first discharge [24]. This report is a motivation for further study on electrochemical performance of this material. For example, the capacity retention of 800 mAh g−1 was obtained in the fabrication of Ca3Co4O9 thin films as anode for LIBs after 50 cycles [25]. Furthermore, the comparison of morphologies, i.e., nanoparticles, multilayer structure, and micro-plates on electrochemical properties of this material, was investigated [26]. It was found that the electrochemical performance of nanoparticle was higher than that of the multilayer and micro-plates due to the short of lithium diffusion transport [26]. In addition, the improved electrochemical performance of Ca3Co4O9 could be done by doping other metals in the system. For example, Ko et al. showed the iron-substituted calcium cobaltite (Ca3Co3FeO9) as an applicable anode material with outstanding rate capability [27]. The in situ formation of active/inactive nanocomposites improved the conversion reaction kinetics by accommodating the large volume change during lithiation/delithiation process [27]. Moreover, potassium doping (Ca2.95K0.05Co4O9) could improve the capacity and stability almost twice compared to the undoped sample [28]. Recently, the effect of chromium doping (Ca3Co3.9Cr0.1O9) on the electrochemical performance was investigated [29]. It was found that Cr doping could enhance the reactivity, cycling performance, and rate capability of Ca3Co4O9 [29]. In this work, the Co atom in this system was replaced by cheaper and more eco-friendly of Fe and/or Mn atoms. Hence, Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 were synthesized and investigated on their electrochemical properties. The effect of transition metal (Fe or Mn) doping was discussed.
Experimental
The Ca3Co4O9, Ca3Co3.9Fe0.1O9 and Ca3Co3.9Mn0.1O9 powders were synthesized from the solution of acetate salts by a thermal hydro-decomposition method. High purities of calcium acetate (Poch, 99%), cobalt acetate (American Element, 99.9%), iron acetate (Aldrich, 99.99%), and manganese acetate (Aldrich, 99.99%) were employed as starting materials without further purification. In the synthesized procedure, the metal salts were dissolved in the stoichiometric ratios in de-ionized (DI) water. The ratio of the DI water to total metal salts was 5 ml/1 g. The mixed solution was stirred with a magnetic stirrer until completely dissolved at room temperature. After that, the solution was calcined at 1053 K for 6 h and then at 1123 K for 24 h in a furnace under normal atmosphere.
The phases of all synthesized samples were identified by X-ray diffraction (XRD, Bruker, D8 Advance) using Cu-K α radiation (wave length = 1.5406 Å) over 2θ range of 5–65°. The chemical compositions were verified by the energy dispersive X-ray spectroscopy (EDS) and the proton induced X-ray emission (PIXE) techniques using a 2.0 MeV proton beam. The morphology of the powders was studied using a field emission scanning electron microscope (FE-SEM, JEOL, JSM 7600F).
For the electrochemical experiments, the working electrode of a coin-type cell was prepared as described in Ref. 29. The active mass (Ca3Co4O9, Ca3Co3.9Fe0.1O9 or Ca3Co3.9Mn0.1O9) powders were mixed with carbon black (CB) and polyvinylidene fluoride (PVDF) binder in 1-methyl-2-pyrrolidinone (NMP) by which the weight ratio of the active mass/CB/PVDF was 67:20:13. To make the slurry, the mixture was continuously stirred with a magnetic stirrer for 48 h. Then, the obtained slurry was cast onto a copper foil followed by an evaporation of the solvent and dried at 333 K in a vacuum oven overnight. Cell assembling and sealing were carried out in an argon-filled glove box, where both moisture and oxygen levels were less than 0.1 ppm. The weight of active material in each electrode was used in amount of 0.6–0.7 mg. The lithium foils were used as the counter/reference electrodes. The electrolyte was the solution of 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1/1, v/v). The assembled cells were galvanostatically cycled using a NEWARE multi-channel battery test system in the voltage range of 0.001–3.0 V with the rate ranging from 0.1 to 5 C at room temperature. The cyclic voltammetry (CV) measurements were carried out with a scan rate of 0.2 mV s−1 in the potential range of 0.005–3.0 V using a Solartron electrochemical instrument potentiostat (SOL-1470E, Solartron, UK).
Results and discussion
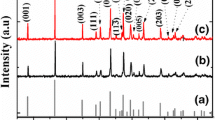
The XRD patterns of the Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 powders are shown in Fig. 1. All diffraction peaks of each sample match well with the standard card (PDF 21-0139) and are consistent with the Ca3Co4O9 structure without any impurity phase. The lattice planes are identified according to the previous studies [16]. This shows that both Fe and Mn doping do not change the crystal structure of Ca3Co4O9. The inset of Fig. 1 shows the close up of the (002) peak which is shifted to the lower angle when transition metals are doped. Hence, it is expected that the dopants were substituted at the Co-sites in the structure. The shift to the lower angle of the XRD peaks for the Fe or Mn doping is attributed to substitution of bigger ions. In previous reports, the charge states of Co, Fe, and Mn ions in this system were found to be +2/+3/+4, +2/+3, and +3, respectively [19, 30]. According to Shannon’s table, the ionic radii for Co, Fe, and Mn in six-coordination both high spin state (HS) and low spin stat (LS) are Co2+ (0.745 Å HS, 0.55 Å LS), Co3+ (0.61 Å HS, 0.545 Å LS), Co4+ (0.53 Å HS), Fe2+ (0.78 Å HS, 0.61 Å LS), Fe3+ (0.645 Å HS, 0.55 Å LS), and Mn3+ (0.645 Å HS, 0.58 Å LS) [31]. In previous report, the spin states of Co were reported as low spin state configuration [30]. However, the spin state of Fe and Mn ions in the Ca3Co3.9M0.1O9 (M = Fe or Mn) system is very difficult to determine. The previous studies of Ca3Co3.9M0.1O9 (M = Fe or Mn) did not show any evidence for the spin state of Fe ions or Mn ions [19, 30, 32]. Considering the ionic radius of Fe2+/Fe3+ and Mn3+ in both low spin state and high spin state are slightly bigger than that of Co ion in low spin state. Thus, partially substituting Fe or Mn for Co ions resulted in the increment of lattice parameters leading to the shift to the lower angel in Fig. 1. The SEM images of the samples are shown in Fig. 2. It could be seen that all samples exhibited the morphology of plate-like particles. The partial substitutions of Fe or Mn at Co-sites of Ca3Co4O9 do not change on the surface morphology. The average particle size of all samples was ∼1–2 μm. The particle size of the doped samples was slightly larger than that of the undoped one. The possible reason is an increment of lattice parameters of the doped samples. To further confirm phase identification, the quantitative analysis of the EDS and PIXE spectra are illustrated in Table 1. These results show that the chemical compositions of each sample are closed to the nominal composition.

X-ray diffraction patterns of a the standard reference (PDF 21-039), b Ca3Co4O9, c Ca3Co3.9Fe0.1O9, and d Ca3Co3.9Mn0.1O9. The inset expands the (002) peak demonstrating the shift to the lower angle with transition metal doping

SEM micrographs of the calcined powders: a Ca3Co4O9, b Ca3Co3.9Fe0.1O9, and c Ca3Co3.9Mn0.1O9
To investigate the effect of transition metal (Fe or Mn) doping on electrochemical properties of Ca3Co4O9, the cobaltite samples based on the half-cell configuration were determined by the cyclic voltammetry (CV) and galvanostatic cycling. Figure 3 shows the CV profiles of 1st–5th cycles of the Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 negative electrodes between 0.005 and 3.0 V at a scan rate of 0.2 mV s−1. In the first cycle, all samples exhibited the prominent reduction peak at 0.4 V and the broad oxidation peak located at 2.0 V. This reduction peak could be assigned to the crystal structure destruction process [33] and the reaction of lithium with the active mass to form transition metals (Co, Fe, or Mn), Li2O, and inactive CaO. In this oxide, an inactive CaO existed after full lithiation because it cannot be reduced to metallic Ca due to its high bond strength [24], which differs from the other oxides; all of cations were reduced to metal in the first discharge reaction, such as MnCo2O4 and CoMn2O4 [34, 35]. On the other hand, the oxidation peak could be attributed to the reaction of metals to metal oxides, for example, Co to CoO and/or Co3O4 [24]. In the subsequent cycles, the reduction peak was shifted to 0.6 V. This is due to the reduction of polarization induced by smaller size of electrognerated nanograins [36]. It could be attributed to the reaction of lithium with the metal oxides such as CoO, Fe3O4, and MnO to generate metallic species (Co, Fe, or Mn), Li2O, and inactive CaO compound. In addition, it could be clearly seen that the intensity of oxidation and reduction peaks decreased with an increase of cycle number for undoped sample as shown in Fig. 3a. It could be referred to the irreversible capacity of the electrode corresponding to a small capacity fading.

The cyclic voltammogram at a scanning rate of 0.2 mV s−1 of the following: a Ca3Co4O9, b Ca3Co3.9Fe0.1O9, and c Ca3Co3.9Mn0.1O9
Figure 3b, c shows the CV profiles of the doped samples. The profiles almost overlapped in the subsequent cycles excepted for the second cycle, indicating good reversibility of the electrodes. A slight difference in the second cycle might be resulted from an incomplete reaction of lithium with metal oxides. In addition, to compare the CV profiles quantitatively, the areas under the oxidation and reduction peaks are evaluated as shown in Table 2. It could be clearly seen that the doped samples showed much higher peak areas than those of undoped sample. This indicates the higher reactivity of the Ca3Co3.9Fe0.1O9 and Ca3Co3.9Mn0.1O9 electrodes which could contribute to the improvement of capacity. The peak areas of Ca3Co3.9Fe0.1O9 were slightly higher than those of Ca3Co3.9Mn0.1O9, indicating higher capacity.
In the galvanostatic mode, the charge/discharge cycling of the Ca3Co4O9, Ca3Co3.9Fe0.1O9 and Ca3Co3.9Mn0.1O9 negative electrodes carried out in the potential window of 0.001–3.0 V at a rate of 0.1 C is shown in Fig. 4. Figure 4a shows the first and 50th charge/discharge profiles. All samples exhibited similar three regions in the first discharge voltage curve. In the first region, the voltage abruptly dropped from 3.0 to 0.9 V. This region refers to the insertion of a very small amount of lithium-ions into the crystal structure [7, 24]. It can be seen that the insertion of lithium-ions in Ca3Co4O9 was larger than that of Ca3Co3.9Fe0.1O9 and Ca3Co3.9Mn0.1O9, corresponding to the higher initial capacity of 200 mAh g−1 for Ca3Co4O9 and of 120 mAh g−1 for the doped samples. This might be due to the slightly smaller particle of Ca3Co4O9 which resulted in higher surface area. The second region was the long plateau with the voltage of 0.9 V. The last region was the gradual voltage decrease to 0.001 V. The first discharge capacities of ∼1000 mAh g−1 were obtained for all samples. These values were higher than their theoretical capacities (∼643 mAh g−1), which was an effect of the solid electrolyte interface (SEI) layer as discussed in the previous reports [24, 29]. This suggests a similar electrochemical process in the first discharge. Hence, the first discharge reaction led to the irreversible crystal destruction and the formation of transition metals (Co, Fe, or Mn) dispersed in the Li2O and inactive CaO which could be written as follows:

The galvanostatic charge/discharge capacities at a rate 0.1 C of Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9. a The first (solid line) and 50th (dashed line) cycles. The subsequent cycles are shown in b Ca3Co4O9, c Ca3Co3.9Fe0.1O9, and d Ca3Co3.9Mn0.1O9
During the first charge, all samples exhibited an analogous charge profiles, indicating similar electrochemical reactions. The charge profile had no plateau and the slope changed at ∼2.0 V. The undoped sample exhibited a capacity of 397 mAh g−1 while the doped samples showed higher capacities of 537 and 518 mAh g−1 for Ca3Co3.9Fe0.1O9 and Ca3Co3.9Mn0.1O9, respectively. The capacity of Ca3Co3.9Fe0.1O9 was slightly higher than that of Ca3Co3.9Mn0.1O9, as the above discussion, due to its high reactivity. In addition, during the first charge/discharge, the undoped sample showed much higher capacity loss compared to the doped samples. The losses of capacities are a result of irreversible components during the first charge/discharge cycle; 59% for Ca3Co4O9, 48% for Ca3Co3.9Fe0.1O9, and 50% for Ca3Co3.9Mn0.1O9. This could also be observed in other metal oxide electrodes [3, 4, 37]. In the first charge reaction, the transition metals (Co, Fe, or Mn) were converted to their oxides. On the other hand, their oxides were also reduced to their metals in the subsequent discharge cycles. The charge/discharge reaction can be expressed as follows:
where A, B, C, x, and y are real numbers which balance the equation.
Figure 4b–d shows the specific capacities of selected charge/discharge profiles in the subsequent cycles for clarity. For the charge profiles, all sample showed the similar curve as the first charge profile. It has no plateau and changes the slope at voltage of 2.0 V. This indicates the same oxidation reaction as the first charge reaction. The specific charge capacities of the doped samples were gradually to increase with an increase of cycling number while the capacity of the undoped sample tended to decrease from the 2nd cycle to 40th cycle and seem to increase slightly in the 50th cycle. The discharge voltages could be divided into two regions for all samples. The first region was the abrupt decrease of voltage from 3.0 to 1.2 V. The slope profiles were likely to decrease with an increase of cycles. In this region, the capacities of the undoped and doped samples were obtained as 25 and 80 mAh g−1, respectively. The characteristic of this region was also observed in the previous systems such as Ca3Co4O9 [24], Ca3Co3Fe1O9 [27], and Co3O4 [7]. It was referred to the formation of intermediate compounds, for instance, LixCo3O4 or CoO, which were decomposed to Co and Li2O for further reduction [7, 24, 27]. The second region was the gradual decrease of the voltage from 1.2 to 0.001 V. The specific discharge capacities of the undoped sample gradually decreased with the number of cycles as shown in Fig. 4b, which is about 13.4% decrease. On the other hand, Fig. 4c, d shows the stable charge/discharge capacities of the doped samples over the cycling measurement. This indicates that the partial Fe or Mn doping improved the cycling performance of Ca3Co4O9. In addition, the capacity of Ca3Co3.9Mn0.1O9 was slightly higher than that of Ca3Co3.9Fe0.1O9 after charge/discharge of 10 cycles. This indicates that the reactivity of Ca3Co3.9Mn0.1O9 might be enhanced during subsequent charge/discharge process. The specific discharge capacities of Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 samples at the 50th cycle were 348, 562, and 581 mAh g−1, respectively. It could be clearly seen that the capacities of the doped samples were much larger than those of the undoped sample, with an increase of over 60%. When compared between the dopants (Fe and Mn), the difference in the capacity is not significant in the present study.
Our results showed that the substituted transition metals could improve the capacity and stability in the Ca3Co4O9 system as anode electrodes for LIBs. The effect of Fe or Mn doping is a significant factor to improved electrochemical performance. The possible reasons for an improvement of capacity in the doped samples might be due to a few possible contributions. The first contribution is the metal oxide-lithium conversion reaction. In case of Fe doping, the iron oxide–lithium (Fe3O4/Li) conversion reaction is a possible reason for enhanced capacity of Ca3Co3.9Fe0.1O9/Li cell because the approximate voltage of this reaction is similar to the present study [37]. On the other hand, based on the manganese oxide compounds Mn3O4, Mn2O3, and MnO2, their oxide–lithium ion (MxOy/Li) is much too low to be a possible reason for enhanced capacity of Ca3Co3.9Mn0.1O9/Li cell. The MxOy/Li conversion reaction exhibited the approximate voltage of only ∼0.4 V [38–40]. The exception is MnO in the MnO/Li conversion reaction. The discharge profile of MnO/Li cell exhibits the voltage plateau of 0.6 V [3], similar to the present study. Hence, MnO/Li conversion reaction could be a possible reason for the enhanced capacity of Ca3Co3.9Mn0.1O9/Li cell. Furthermore, another contribution is the electrochemical reaction of electrode materials. In both cases (Fe and Mn doping), the reduction of volume change could mainly contribute to the improvement of the capacity. Co and Fe metals preferred the oxidation states as CoO and Fe3O4 [27] and the most stable oxidation of MnO was expected. The Ca3Co3.9Fe0.1O9/Li and Ca3Co3.9Mn0.1O9/Li conversion reactions and volume change could be determined as illustrated in Table 3. The detail of the calculation is available in the supporting data. The Fe or Mn doping reduced the volume change and maintained high theoretical capacity. As shown in Table 3, the volume changes in the first discharge reaction were reduced to ∼55%, and further reduced to 24.5%, and 23.6% in the subsequent cycles for Ca3Co3.9Fe0.1O9 and Ca3Co3.9Mn0.1O9, respectively.
The galvanostatic charge/discharge cycling results at the rate of 0.1, 0.2, 0.5, 1, 2, and 5 C are shown in Fig. 5. The doped samples exhibited the higher charge/discharge capacities over all range of measurements, indicating a high rate capability. However, the capacity of Ca3Co3.9Fe0.1O9/Li cell was rapidly decreased at a rate of 5 C. This indicates that the capacity of Ca3Co3.9Mn0.1O9/Li cell was better than that of the Ca3Co3.9Fe0.1O9/Li cell in the high C-rate. In addition, the coulombic efficiency was higher 93%, indicating a high ratio of delithiation/lithiation.

The galvanostatic charge/discharge capacities and efficiency of Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 at the rate of 0.1, 0.2, 0.5, 1, 2, and 5 C. The half-filled, hollow, and filled symbols represent efficiency, charge, and discharge, respectively
Conclusions
Ca3Co4O9, Ca3Co3.9Fe0.1O9, and Ca3Co3.9Mn0.1O9 powders were successfully synthesized and tested for anode electrode candidate in LIBs. The transition metal (Fe or Mn) doping could improve the cycling performance and rate capability of Ca3Co4O9. The enhancement of capacity resulted from the higher reactivity, the reduction in volume change, and the transition metal oxide–lithium conversion reaction. In the redox reactions, the Co, Fe, and Mn metals were likely to be oxidized as CoO, Fe3O4, and MnO, respectively. Further studies for the research on other metal doping, such as Ni, Cu, and Zn, are likely to be the key to improve the capacity of this anode material.
References
Wang F, Wang J, Ren H, Tang H, Yu R, Wang D (2013) Multi-shelled LiMn2O4 hollow microspheres as superior cathode materials for lithium-ion batteries. Inorg Chem Front 3:265
Scrosati B (2000) Recent advances in lithium ion battery materials. Electrochim Acta 45:2461
Li X, Shang X, Li D, Yue H, Wang S, Qiao L, He D (2014) Facile synthesis of porous MnO microspheres for high-performance lithium-ion batteries. Part Part Syst Charact 31:1001
Poizot P, Laruelle S, Grugeon S, Dupont L, Tarascon J-M (2000) Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries. Nature 407:496
Taberna PL, Mitra S, Poizot P, Simon P, Tarason J-M (2006) High rate capabilities Fe3O4-based Cu nano-architectured electrodes for lithium-ion battery applications. Nat Mater 5:567
Li X, Li D, Qiao L, Wang X, Sun X, Wang P, He D (2012) Interconnected porous MnO nanoflakes for high-performance lithium ion battery anodes. J Mater Chem 22:9189
Reddy MV, Prithvi G, Loh KP, Chowdari BVR (2014) Li storage and impedance spectroscopy studies on Co3O4, CoO, and CoN for Li-ion batteries. Appl Mater Interfaces 6:680
Xiao Y, Li X, Zai J, Wang K, Gong Y, Li B, Han Q, Qian X (2014) CoFe2O4-graphene nanocomposites synthesized through an ultrasonic method with enhanced performances as anode materials for Li-ion batteries. Nano Micro Lett 6:307
Jiang S, Shi T, Long H, Sun Y, Zhou W, Tang Z (2014) High-performance binder-free supercapacitor electrode by direct growth of cobalt-manganese composite oxide nansostructures on nickel foam. Nanoscale Res Lett 9:492
Wang J, Tang H, Zhang L, Ren H, Yu R, Jin Q, Qi J, Mao D, Yang M, Wang Y, Liu P, Zhang Y, Wen Y, Gu L, Ma G, Su Z, Tang Z, Zhao H, Wang D (2016) Multi-shelled metal oxides prepared via an anion-adsorption mechanism for lithium-ion batteries. Nat Energy 1:16050
Wang J, Yang N, Tang H, Dong Z, Jin Q, Yang M, Kisailus D, Zhao H, Tang Z, Wang D (2013) Accurate control of multishelled Co3O4 hollow microspheres as high-performance anode materials in lithium-ion batteries. Angew Chem Int Ed 52:1
Xu S, Hessel CM, Ren H, Yu R, Jin Q, Yang M, Zhao H, Wang D (2014) α-Fe2O3 multi-shelled hollow microspheres for lithium ion battery anodes with superior capacity and charge retention. Energy Environ Sci 7:632
Ren H, Yu R, Wang J, Jin Q, Yang M, Mao D, Kisailus D, Zhao H, Wang D (2014) Multishelled TiO2 hollow microspheres as anodes with superior revesible capacity for lithium ion batteries. Nano Lett 14:6679
Ren H, Sun J, Yu R, Yang M, Gu L, Liu P, Zhao H, Kisailus D, Wang D (2016) Controllable synthesis of mesostructures from TiO2 hollow to porous nanospheres with superior rate performance for lithium ion batteries. Chem Sci 7:793
Qi J, Lai X, Wang J, Tang H, Ren H, Yu Y, Jin Q, Zhang L, Yu R, Ma G, Su Z, Zhao H, Wang D (2015) Multi-shelled hollow micro-/nanostructrues. Chem Soc Rev 44:6749
Masset AC, Michel C, Maignan A, Hervieu M, Toulemonde O, Studer F, Raveau B, Hejtmanek J (2000) Misfit-layered cobaltite with an anisotropic giant magnetoresistance: Ca3Co4O9. Phys Rev B 62:166
Nong NV, Pryds N, Linderoth S, Ohtaki M (2011) Enhancement of the thermoelectric performance of p-type layered oxide Ca3Co4O9+δ through heavy doping and metallic nanoinclusions. Adv Mater 23:2484
Prasoetsopha N, Pinitsoontorn S, Amornkitbamrung V (2012) Synthesis and thermoelectric properties of Ca3Co4O9 prepared by a simple thermal hydro-decomposition method. Electron Mater Lett 8:305
Prasoetsopha N, Pinitsoontorn S, Bootchanont A, Kidlhunthod P, Srepusharawoot P, Khamwanna T, Amornkitbamrung V, Kurosaki K, Yamanaka S (2013) Local structure of Fe in Fe-doped misfit-layered calcium cobaltite: An X-ray absorption spectroscopy study. J Solid State Chem 204:257
Miyazaki Y (2004) Crystal structure and thermoelectric properties of the misfit- layered cobalt oxides. Solid State Ionics 172:463
Rebola A, Klie RF, Zapol P, Ogut S (2012) Lattice and transport properties of the misfit-layered oxide thermoelectric Ca3Co4O9 from first principles. Phys Rev B 85:155132
Pinitsoontorn S, Lerssongkram N, Harnwunggmoung A, Kurosaki K, Yamanaka S (2010) Synthesis, mechanical and magnetic properties of transition metals-doped Ca3Co3.8M0.2O9. J Alloys Compd 503:431
Prasoetsopha N, Pinitsoontorn S, Thongbai P, Yamwong T (2013) Giant dielectric behavior observed in Ca3Co4O9 ceramic. Electron Mater Lett 9:347
Kim D-W, Ko Y-D, Park J-G, Kim B-K (2007) Formation of lithium driven active/inactive nanocomposite electrodes based on Ca3Co4O9 nanoplates. Angew Chem Int Ed 46:6654
Zhu X-B, Chou S-L, Wang L, Li Q, Shi D-Q, Wang J-Z, Chen Z-X, Sun Y-P, Liu H-K, Doua S-X (2009) Self-oriented Ca3Co4O9 thin film as an anode material for enhanced cycling stability of lithium-ion batteries. Electrochem Solid State Lett 12:A176
Mei T, Zhang J, Wang L, Xing Z, Zhu Y, Qian Y (2012) Preparation of mixed oxides Ca9Co12O28 and their eletrochemical properties. Mater Lett 82:1
Ko Y-D, Kang J-G, Choi KJ, Park J-G, Ahn J-P, Chung KY, Nam K-W, Yoon W-S, Kim D-W (2009) High rate capabilities induced by multi-phasic nanodomains in iron-substituted calcium cobaltite electrodes. J Mater Chem 19:1829
Cao J, Liu H, Xie J, Cao G, Zhao X (2010) Effect of K-doping on the electrochemical performance of Ca3Co4O9 anode for Li-ion batteries. J Mater Sci Technol 26:669
Prasoetsopha N, Pinitsoontorn S, Kamwanna T, Meethong N, Fan S, Tan LP, Sun T, Hng HH (2015) Improvement of electrochemical properties of misfit-layered Ca3Co4O9 as anode materials for lithium-ion batteries by Cr doping. J Solid State Electrochem 19:1197
Chen JL, Liu YS, Liu C-J, Huang L-C, Dong CL, Chen SS, Chang CL (2009) Effect of Mn doping on the physical properties of misfit-layered Ca3Co4O9+δ. J Phys D App Phys 42:135418
Shannon R (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chaleogenides. Acta Crystallogr A 32:751
Liu CJ, Chen JL, Huang LC, Lin ZR, Chang CL (2007) X-ray absorption spectroscopy studies of Fe-doped misfit-layeered Ca3Co4-xFexO9+δ. J Appl Phys 102:014908
Sharma N, Shaju KM, Rao GVS, Chowdari BVR (2004) Mixed oxides Ca2Fe2O5 and Ca2Co2O5 as anode materials for Li-ion batteries. Electrochim Acta 49:1035
Fu C, Li G, Luo D, Huang X, Zheng J, Li L (2014) One-step calcination-free synthesis of multicomponent spinel assembled microspheres for high-performance anodes of li-ion batteries: a case study of MnCo2O4. ACS Appl Mater Interfaces 6:2439
Li G, Xu L, Zhai Y, Hou Y (2015) Fabrication of hierarchical porous MnCo2O4 and CoMn2O4 microspheres composed of polyhedral nanoparticles as promising anodes for long-life LIBs. J Mater Chem A 3:14298
Ge X, Gu CD, Wang XL, Tu JP (2014) Correlation between microstructure and electrochemical behavior of the mesoporous Co3O4 sheet and its ionothermal synthesized hydrotalcite-like α-Co(OH)2 precursor. J Phys Chem C 118:911
Xiong QQ, Tu JP, Lu Y, Chen J, Yu YX, Qiao YQ, Wang XL, Gu CD (2012) Synthesis of hierarchical hollow-structured single-crystalline magnetite (Fe3O4) microspheres: The highly powerful storage versus lithium as an anode for lithium ion batteries. J Phys Chem C 116:6495
Hu L, Sun Y, Zhang F, Chen Q (2013) Facile synthesis of porous Mn2O3 hierarchical microspheres for lithium battery anode with improved lithium storage properties. J Alloys Compd 576:86
Xia H, Laia M, Lu L (2010) Nanoflaky MnO2/carbon nanotube nanocomposites as anode materials for lithium-ion batteries. J Mater Chem 20:6896
Wang H, Cui L-F, Yang Y, Casalongue HS, Robinson JT, Liang Y, Cui Y, Dai H (2010) Mn3O4-graphene hybrid as a high-capacity anode material for lithium ion batteries. J Am Chem Soc 132:13978
Acknowledgements
This work is supported by Suranaree University of Technology (SUT) and by the Office of the Higher Education Commission under NRU project of Thailand, the Thailand Research Fund (TRF) in cooperation with the Commission on Higher Education and Khon Kaen University (Grant No. MRG5680159) and the Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Center of Excellence Network.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOC 43 kb)
Rights and permissions
About this article

Cite this article
Prasoetsopha, N., Pinitsoontorn, S., Fan, S. et al. Enhancement of electrochemical properties of Ca3Co4O9 as anode materials for lithium-ion batteries by transition metal doping. Ionics 23, 395–403 (2017). https://doi.org/10.1007/s11581-016-1943-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-016-1943-3