
Abstract
The LiFePO4/C composite with a grape bunch structure was synthesized through the hydrothermal method at 170 °C for 7 h and followed by being fired at 750 °C for 4 h. Commercial Li2CO3, (NH4)2Fe(SO4)2 · 6H2O, and (NH4)2HPO4 were used as raw materials. Glucose was used as in situ coating carbon source, and the hydroxylated MWCNTs were used as connecting carbon wires which could be embedded into the carbon coating to form a uniform grape bunch structure. The resultant samples were characterized by X-ray diffraction (XRD), scanning electron microscope (SEM), transmission electron microscopy (TEM), energy dispersive spectrometry (EDS), elementary analysis (EA), Raman spectroscopy, and electrochemical tests. The results show that the grape bunch structure with a low disordered/graphene (D/G) ratio was found to be well dispersed in the LiFePO4/C composite, and a three-dimensional carbonaceous network was formed which could enhance the electronic conductivity of the LiFePO4/C composite remarkably. The resultant LiFePO4/C composite shows a high discharge capacity of 160.3 mAh g−1 at 0.1 C and 110.9 mAh g−1 even at 10 C, and the cycling capacity retention rate reaches 99.6 % over 60 cycles. Besides, it also exhibits high conductivity, good reversibility, and excellent stability in EIS and CV tests.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the pioneering work of Padhi et al. in 1997 [1], the phospho-olivine LiFePO4 has been recognized as one of the most promising cathode materials for rechargeable Li-ion batteries, owing to its low cost, high theoretical capacity (170 mAh g−1), improved safety, and high stability [2, 3]. However, the low ionic and electronic conductivity has become the main obstacles for its commercial applications [4, 5]. Tremendous efforts have been made to enhance the conductivity of LiFePO4 by material modifications and adopting various synthesis methods in the past decade [6, 7]. In all these methods, carbon coating is the most popular method for improving the electronic conductivity of LiFePO4 [8, 9]. These composites are typically prepared by adding organic or polymeric precursors during synthesis, and the conductivities of these composites are largely depended upon the carbon structure and the carbon dispersion degree [10, 11]. Optimization of the carbon structure, particularly the sp2/sp3 and disordered/graphene (D/G) ratios, could improve the electronic conductivity remarkably [12, 13]. Meanwhile, the manipulation of the carbon structure can be achieved via using various kinds of the carbon precursors, adding graphitization catalysts, and optimizing the carbonization conditions [14, 15]. Carbon nanotubes (CNTs) are one of the most promising carbon materials due to their relatively high electrical conductivity, large surface area, and low D/G ratio [16, 17].
Generally, LiFePO4 is typically prepared by various synthetic techniques such as solid-state, sol-gel, and hydrothermal method [18–20]. Among them, hydrothermal method arouses particular interest due to its mild operating temperature, simple process, and the potential for large-scale production [21, 22]. Up to now, there have been many reports about the effects of particle size, crystallographic structure, and electronic conductivity on the electrochemical performance of LiFePO4 by the hydrothermal method. Lee and Teja [23] investigated the effects of hydrothermal synthesis conditions, such as temperature, reactant concentration, pH value, and reaction time, on the particle size, purity, and capacity of LiFePO4. Dokko et al. [24, 25] synthesized LiFePO4 through a hydrothermal process at 170 °C for 12 h, followed by annealing at 400 °C for 30 min in an argon atmosphere. The prepared sample (carbon free), with a particle size of 0.5 μm, exhibited a discharge capacity of 150 mAh g−1 at 0.2 C, 80 mAh g−1 at 0.5 C, and 40 mAh g−1 at 5 C in a potential range of 2.0–4.5 V. Meligrana et al. [26] developed a hydrothermal synthesis to prepare LiFePO4/C composite in the presence of cetyltrimethylammonium bromide (CTAB), with a high concentration at 120 °C for 5 h, then fired at 600 °C for 12 h in pure N2. The conductivity of the composite was improved by the in situ coated carbon film on the surface of the particles. The sample with 4.8 wt% carbon showed discharge capacities of 140 mAh g−1 at 0.2 C and 115 mAh g−1 at 5 C. However, the electrochemical performance (practically the high-rate capacity) of LiFePO4 prepared by the hydrothermal method reported so far is not up to expectation. It is believed that optimization of the conducting phase coating on LiFePO4 particles prepared through hydrothermal method is the important thing to improve electrochemical performances of LiFePO4 [27, 28].
Recently, we successfully synthesized spherical LiFePO4/C composite with a carbon coating via a novel glucose-assisted hydrothermal method, and the inexpensive Li2CO3 was used as lithium source [29, 30]. In order to further improve the electrochemical performance of the LiFePO4/C composite, we employ the hydroxylated multiwalled CNTs (MWCNTs, a kind of CNTs which could be well dispersed in aqueous solution) as connecting carbon wires to link the carbon coating on the LiFePO4 microspheres, and the hydroxylated MWCNTs could be embedded in the carbon coating to form a grape bunch structure. The grape bunch structure owing to its three-dimensional conductive network and low D/G ratio could greatly increase the contacting area and conductivity of carbonaceous material comparing with the “plot to plot” contacting of the carbon coating. Here, we present the preparation, structure, morphology, and electrochemical performances of LiFePO4/C cathode for Li-ion batteries.
Experimental
The LiFePO4/C powder was produced through a novel hydrothermal method. Starting materials were Li2CO3(A.R.), (NH4)2Fe(SO4)2 · 6H2O(A.R.), (NH4)2HPO4(A.R.), glucose(A.R.), and hydroxylated MWCNTs (purity of 95 %, Heluelida Power Material Co. Ltd. Xinxiang, China). The molar ratio of n Li:n Fe:n P was controlled at 1:1:1. First, (NH4)2Fe(SO4)2 · 6H2O and (NH4)2HPO4 were dissolved in 200-mL deionized water separately, and glucose was also added into the (NH4)2Fe(SO4)2 · 6H2O solution. The concentration of Fe2+ and the glucose in the (NH4)2Fe(SO4)2 · 6H2O solution was controlled to be 0.625 and 0.15 mol L−1, respectively. Then, the two kinds of solution were mixed with a fixed sequence under the magnetic stirring in a beaker containing Li2CO3 powder, and the beaker was heated in a boiling water bath. A certain concentration (3 g L−1) of hydroxylated MWCNT aqueous solution (100 mL), which was prepared by ultrasonic dispersing in 50 °C water bath for 50 min, was also added to the beaker under continuous stirring. Meanwhile, the pH value of the solution system was adjusted to a set value (pH = 7) using ammonia solution. The resulting gray slurry was transferred into a 1-L stainless steel autoclave equipped with a mechanical stirrer (Weihai Co., model GSH-1, China), and the autoclave was purged with nitrogen to remove residual oxygen. Then, the autoclave was sealed and heated at 170 °C for 7 h. After the hydrothermal reaction, the autoclave was cooled at room temperature. Afterward, the precipitate was filtered and washed with deionized water and absolute ethanol separately, then dried in a vacuum oven at 120 °C for 4 h. In order to further carbonize the carbonaceous products obtained by hydrothermal carbonization of glucose, the dried precipitate was subsequently two-step carbonized in a tube furnace at 500 °C for 1 h and then 750 °C for 4 h under an N2 (99.99 %) flow. After cooling, the final black-colored LiFePO4/C composites were obtained. The samples were hereafter reported as LFP/C. For comparison, LiFePO4 composites were also separately prepared with MWCNTs and the absence of glucose (hereafter reported as LFP/CNTs) and with glucose and the absence of the hydroxylated MWCNTs (hereafter reported as LFP/G) under the same conditions.
The structure and morphology of the samples were characterized with X-ray diffractometer (XRD, Philips X’pert Pro MPD) with a CuKα radiation (λ = 1.54178 Å), scanning electron microscope (SEM, Jeol JEM-5900LV), and transmission electron microscope (TEM, Tecnai G2 F20 S-TWIN). The lattice parameters of the samples were calculated by the software of Jade 5.0. The special surface area of the samples was determined by using a BET surface area analyzer (Micromeritics Gemini VII). The structural variations and the dispersion of carbonaceous material in the composites were identified by Raman spectroscopy (HORIBA LabRAM HR, 633-nm HeNe laser) and an energy dispersive spectrophotometer (EDS, X-Max 51-XMX0019). The elemental compositions in the LiFePO4/C composites were determined by an inductively coupled plasma optical emission spectrometer (ICP-OES, SPECTRO ARCOS). The weight percentages of carbon in the LiFePO4/C composites were determined by an elemental analyzer (EA, Italy Euro EA 3000).
The electrochemical performances of the samples were investigated via CR2032 coin-type model cells assembled in an argon-filled glove box. Each cell was composed of a lithium anode and a LiFePO4/C cathode. The cathode was prepared by blending the LiFePO4/C composite, acetylene black, and the binder (7.5 wt% polyvinylidene fluoride in N-methyl-2-pyrrolidinone) at the weight of 80:15:5; then, the mixture was rolled into a thin sheet with uniform thickness on the aluminum foil and cut to a fixed size to fit the CR2032 shell. The used electrolyte was 1 mol L−1 LiPF6 in a 1:1 mixture of ethylene carbonate (EC) and diethyl carbonate (DEC). Celegard 2700 membrane was used as the separator. The galvanostatic cycling tests were carried on a battery test system (Newell, China) with a voltage cutoff of 4.3–2.5 V at room temperature. The electrochemical impedance spectra (EIS) and cyclic voltammetry curves (CVs) were tested on the electrochemical workstation (CHI660C, China).
Results and discussion
XRD characterization
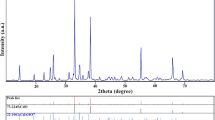
Figure 1 shows the XRD patterns of the samples LFP/CNTs, LFP/G, and LFP/C. It is seen that all diffraction peaks of the three samples can be indexed to an olivine LiFePO4 with orthorhombic crystal structure (JCPDF 40–1499) and there are no impure peaks. All the three samples have narrow diffraction peaks, indicating a high crystallinity degree. Besides, element analysis results show that the carbon contents are 1.58, 2.05, and 3.31 wt% for LFP/CNTs, LFP/G, and LFP/C (listed in Table 1), respectively. However, there are no corresponding diffraction peaks of carbonaceous or carbon phase at detectable levels, probably due to its low content or amorphous state. The lattice parameters of all samples calculated by the software of Jade 5.0 are also listed in Table 1. As being seen, the values of the lattice parameters match well with the standard orthorhombic LiFePO4, certifying that all the three samples have the relatively high purity.

XRD patterns of LiFePO4/C synthesized with different carbon sources: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose
Electron microscopy characterization
SEM images of LFP/G, LFP/CNTs, and LFP/C are presented in Fig. 2. In Fig. 2a, the sample of LFP/G synthesized in glucose solution without hydroxylated MWCNT adding consisted of spherical particles (regarded as the subsequent “grapes”), and the particle size is about 300–350 nm. Figure 2b shows the sample of LFP/CNTs synthesized with hydroxylated MWCNTs without glucose adding; it can be found that the particles are largely cubic morphology and the particle size is 400–450 nm. Moreover, we can find that the hydroxylated MWCNTs (regarded as the subsequent “stalks of grapes”) insert in the cubic particles and the “network-like” structures appear. When the glucose and hydroxylated MWCNTs were both added to synthesize the sample of LFP/C, as shown in Fig. 2c, the hydroxylated MWCNTs (stalks of grapes) were well dispersed in the composite and embed in the sphere particles (grapes) forming a grape bunch structure. The sketch of grape bunch structure model is shown in Fig. 2d. The special surface area of the three samples (LFP/CNTs, LFP/G, and LFP/C) is 6.899, 5.485, and 7.891 m2 g−1, respectively. The surface area of the LFP/CNTs and LFP/C samples with MWCNTs added is superior to that of the LFP/G sample without MWCNTs added. So, it is certified that the CNTs are beneficial to increase the special surface area of the LiFePO4 sample. However, as the added amount of MWCNTs is very low (about 1.58 wt%), the increase of the special surface area of the LFP/CNTs and LFP/C samples is not very obvious.

SEM images of a the sample of LFP/G synthesized with glucose, b the sample of LFP/CNTs synthesized with MWCNTs, c the sample of LFP/C synthesized with MWCNTs and glucose, and d model of the grape bunch structure
The TEM images of the samples of LFP/G and LFP/C are shown in Fig. 3. The LiFePO4 crystallites appear as the dark region, and the carbon coating or MWCNTs appear as the light shade represented by the grayish regions in the TEM images. As shown in Fig. 3a, b of the sample LFP/G, the LiFePO4 particle is uniformly surrounded by a thin (with the average thickness of 20–40 nm) amorphous carbon layer to form a “core-shell” structure. As shown in Fig. 3a, b, the carbon coating of the sample LFP/G exists across the entire surface of the LiFePO4 particles, or the LiFePO4 particle is uniformly surrounded by a thin amorphous carbon layer. The images indicate a core-shell structure with a carbon shell (about 20–40 nm) on the surface of LiFePO4 core (about 300–350 nm). In Fig. 3c, d of the sample of LFP/C, we found that there is also a thin carbon coating on the surface of the LiFePO4 particles and the hydroxylated MWCNTs serve as bridges between particles. Well-dispersed MWCNTs insert in the carbon coating to form a grape bunch structure, and the three-dimensional conducting network exists throughout the composite, which would play an important role in enhancing the conductivity of the LFP/C composite. Elemental mapping for “a bunch of grape” of the sample LFP/C analyzed by EDS in Fig. 4 indicates a uniform distribution of Fe, P, and C on the surface of the sample, and elemental analysis of LFP/C indicates that the Fe/P molar ratio is approximately 1:1. It can also be certified by the analysis of the elemental compositions, which was obtained from the ICP-OES tests. From Table 1, we can calculate that the Li/Fe/P molar ratio of the three samples is all approximately 1:1:1, certifying that the purity of the three samples is very high.

TEM images of a, b the sample LFP/G synthesized with glucose and c, d the sample LFP/C synthesized with MWCNTs and glucose

SEM image (a) and EDS dot-mapping for Fe (b), P (c), and C (d) elements of the sample LFP/C synthesized with MWCNTs and glucose
Raman spectroscopy
Figure 5 shows the Raman spectra of LFP/CNTs, LFP/G, and LFP/C. All the three composites show a well-known D-band (disorder-induced phonon mode) in the range of 1,250 ~ 1,450 cm−1 and G-band (graphite band) between 1,550 and 1,660 cm−1. The peak at around 1,350 cm−1 is attributed to the defects and disordered portions of carbon (sp3-coordinated), and the peak at around 1,600 cm−1 is indicative of ordered graphitic crystallites of carbon (sp2-coordinated). The sharp band at 948 cm−1 in Fig. 5 can be attributed to the symmetric PO4 3− stretching vibration of LiFePO4. The relative I D /I G value generally provides a useful index for comparing the degree of crystallinity of various carbon materials, and smaller ratio of I D /I G indicates the higher degree ordering in the carbon material [31]. As shown in Table 2, the sample LFP/C has the lowest ratio of I D /I G of 0.924 among the three samples, certifying the highest degree ordering of carbon material and best conductivity of the LiFePO4/C composite.

Raman spectra of LiFePO4/C synthesized with different carbon sources: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose
Electrochemical studies
The first charge-discharge profiles (at 0.1 C rate, 1 C = 170 mA g−1) and the cycling performances (at 1 C rate) of the three samples (LFP/CNTs, LFP/G, and LFP/C) are compared in Fig. 6. The sample LFP/CNTs exhibits a discharge capacity of 145.9 mAh g−1 with a significant voltage difference between the charge and discharge curves as seen in Fig. 6a. It also exhibits an obvious fluctuation during the initial cycling test as seen in Fig. 6b, and the capacity retention after 60 cycles is 98.8 %. The sample LFP/G exhibits a discharge capacity of 153.2 mAh g−1 as seen in Fig. 6a, and the capacity retention after 60 cycles is 99.1 %. The lower discharge capacities of LFP/CNTs and LFP/G could be due to the poor electronic conductivity. The discharge-charge efficiency in Fig. 6a of the sample LFP/CNTs is slightly higher than 100 %. The phenomenon could be explained that the LFP/CNT cathode is due to lack of lithium in only a slight degree, which could be calculated from the ICP-OES results of LFP/CNTs in Table 1. The sample LFP/C exhibits a high discharge capacity of 160.3 mAh g−1, which is close to the theoretical value of 170 mAh g−1, and there is little difference between the charge and discharge curves as seen in Fig. 6a. It also exhibits excellent cycling performance as seen in Fig. 6b, and the capacity retention after 60 cycles is 99.6 %. The excellent capacity and cyclability could be related to a significant improvement in the ordered structure of carbon and the good electronic conductivity of the grape bunch structure in the LFP/C composite.

Comparisons of the a first charge-discharge profiles (0.1 C) between 2.5 and 4.3 V and b cycling performances (1 C) of the three samples: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose
In order to evaluate the effect of the grape bunch structure on the rate capability of LiFePO4/C composite, the cells were charged and discharged at different rates from 0.1 to 10 C rates stepwise. The discharge curves of the three samples (LFP/G, LFP/CNTs, and LFP/C) at different rates are compared in Fig. 7, and the corresponding cycling performance is shown in Fig. 8. Combining Figs. 7 and 8, it is discovered that the sample of LFP/C reveals the best rate capacity in the three samples. The decreases of the discharge capacity of LFP/C are less than those of the other two samples when the discharging current is increased from 0.1 to 10 C. The sample of LFP/C owns a discharge capacity of 152.0 mAh g−1 at 0.5 C, 148.4 mAh g−1 at 1 C, 140.4 mAh g−1 at 2 C, and 129.8 mAh g−1 at 5 C. Even at 10 C, the discharge capacity of LFP/C is still as high as 110.9 mAh g−1, and the capacity retention is as high as about 62.9 %. Obviously, the sample of LFP/C shows the best rate capacity performance among the three samples, and the grape bunch structure has a significant improvement on the rate capacity of LiFePO4/C composite. However, the discharge platform of LFP/CNTs seems more stable than that of LFP/C and LFP/G in the initial section of discharge curves at high rates as shown in Fig. 7, which means that the polarizations of LFP/C and LFP/G electrodes are more serious than that of LFP/CNTs electrode to some extent. The decrease of LFP/C and LFP/G platforms explained that the carbon coating on the surface of LiFePO4 core distributes nonuniformly in the limited region, which may cause partially uneven distribution of current density on the surface of the electrode at high current density.

Comparison of the discharge curves of LiFePO4/C synthesized with different carbon sources at various current rates: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose

Comparison of the rate capacities of LiFePO4/C synthesized with different carbon sources: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose
Generally, the EIS may be considered as one of the most sensitive tools for the study of differences in the electrode behavior due to material modification [30]. The EIS of the three samples (LFP/G, LFP/CNTs, and LFP/C) are compared in Fig. 9, which were measured in the fully discharged state after cycling. An intercept at the Z′ axis in high frequency corresponds to the resistance of the electrolyte (R e). The semicircle in the high-middle frequency corresponds to the charge-transfer resistance (R ct). The inclined line in the low frequency represents the Warburg impedance (W s), which is attributed to the diffusion of the lithium ions in the LiFePO4 material. A simplified equivalent circuit model is also shown in Fig. 9, which is constructed to analyze the EIS of the samples. The parameters of the equivalent circuit are recorded in Table 3. Figure 10 shows the relationship plot between Z re and reciprocal square root of the angular frequency (ω−0.5) at low-frequency region. This relation is governed by Eq. (1). Also, the lithium-ion diffusion coefficient is calculated using Eq. (2) and recorded in Table 3.

The impedance spectra of LiFePO4/C synthesized with different carbon sources: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose; the equivalent circuit used for fitting the experimental EIS date

The relationship plots between Z′ and ω−1/2 at low-frequency region of LiFePO4/C synthesized with different carbon sources: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose
where R e is the electrolyte resistant, R ct is the charge-transfer resistance, ω is the angular frequency in the low-frequency region, σw is the Warburg impedance coefficient, D is the diffusion coefficient, R is the gas constant, T is the absolute temperature, A is the area of the electrode surface, n is the number of electron transferred per molecule during the intercalation, F is the Faraday constant, and c is the molar concentration of lithium ions.
It is observed that the Warburg impedance coefficient (σw) is 51.29Ω cm2 s−0.5 for sample LFP/C and it has the lowest value in comparison with the other two samples. The lithium-ion diffusion coefficients of the three samples (LFP/CNTs, LFP/G, and LFP/C) are 5.05 × 10−16, 8.91 × 10−16, and 1.16 × 10−14 cm2 s−1, respectively. The charge-transfer resistance (R ct) of LFP/C is the least, and the lithium-ion diffusion coefficient (D) for the sample is the highest among the three samples; besides, the performances of LFP/C are superior to the reported values (such as the D values were 6.07 × 10−15 cm2 s−1 for LiFePO4/C at room temperature) [32]. Therefore, the grape bunch structure is beneficial for the charge and discharge performance of LiFePO4/C composite. Furthermore, the exchange current density (i 0 = RT/nFR ct, n is the number of electron transferred per molecule during the intercalation) of LFP/C is 5.68 × 10−4 mA cm−2 and higher than that of the other two samples, implying much better reversibility.
In addition, the conductivity σ is calculated from the equation (σ = t/R ct A, t is the thickness of the electrode). It can be seen from Table 3 that the sample of LFP/C has the higher conductivity σ (1.01 × 10−3 s cm−1)than that of the other two samples. It proves that the sample of LFP/C with the grape bunch structure has a relatively unobstructed conductive network, and the conductivity of the sample LFP/C is improved remarkably. Figure 11 shows the cyclic VCs of the three samples (LFP/CNTs, LFP/G, and LFP/C). The sample LFP/C shows highest and sharpest current peaks and smallest charge and discharge plateau difference comparing with LFP/CNTs and LFP/G, implying the best electrode reaction kinetics and stability among the three samples. The EIS and CV tests mainly reflect the overall performance of the LiFePO4/C electrode. Therefore, LFP/C with the grape bunch structure is considered the optimal sample in this research.

The cyclic voltammetry curves of LiFePO4/C synthesized with different carbon sources: LFP/CNTs synthesized with MWCNTs, LFP/G synthesized with glucose, and LFP/C synthesized with MWCNTs and glucose
Conclusions
The LiFePO4/C composites with a grape bunch structure were successfully synthesized by hydroxylated MWCNTs and glucose-assisted hydrothermal method. Glucose was used as in situ coated carbon source, and MWCNTs were used as connected carbon wires which could insert in the coated carbon to form a grape bunch structure. The optimal LiFePO4/C sample exhibits the discharge capacities of 160.3 mAh g−1 at 0.1 C and 110.9 mAh g−1 at 10 C rate, and the cycling capacity retention rate could reach 99.6 % over 60 cycles. The EIS and CV results show that the LiFePO4/C sample has the relatively high conductivity, good reversibility, and excellent stability. The excellent electrochemical performance can be correlated to the well-dispersed MWCNTs forming a grape bunch structure, which leads to the formation of the three-dimensional carbonaceous networks and the enhancement of conductibility of LiFePO4/C composite.
References
Padhi AK, Nanjundaswamy KS, Goodenough JB (1997) J Electrochem Soc 144:1188–1194
Hu YS, Guo YG, Dominko R et al (2007) Adv Mater 19:1963–1966
Lee J, Teja AS (2006) Mater Lett 60:2105–2109
Shu HB, Wang XY, Wu Q et al (2012) Eletrochim Acta 76:120–129
Wang FQ, Chen J, Wu MH et al (2013) Ionics 19:451–460
Vujković M, Stojković I, Cvjetićanin N et al (2013) Eletrochim Acta 92:248–256
Yamada A, Chung SC, Hinokuma K (2001) J Electrochem Soc 148:A224–A229
Wilcox JD, Doeff MM, Marcinek M et al (2007) J Electrochem Soc 154:A389–A395
Murugan AV, Muraliganth T, Manthiram A (2008) J Phys Chem C 112:14665–14671
Zaghib K, Shim J, Guerfi A et al (2005) Electrochem Solid-State Lett 8:A207–A210
Xu ZH, Xu L, Lai QY et al (2007) Mater Res Bull 42:883–891
Doeff MM, Hu YQ, Mclarnon F et al (2003) Electrochem Solid-State Lett 6:A207–A209
Gabrisch H, Wilcox JD, Doeff MM (2006) Electrochem Solid-State Lett 9:A360–A363
Huang YH, Park KS, Goodenough JB (2006) J Electrochem Soc 153:A2282–A2286
Doeff MM, Wilcox JD, Yu R et al (2008) J Solid-State Electrochem 12:995–1001
Toprakci O, Toprakci HAK, Ji L et al (2012) Appl Mater Interfaces 4:1273–1280
Ji L, Yao Y, Toprakci O et al (2010) J Power Sources 195:2050–2056
Chen ZH, Dahn JR (2002) J Electrochem Soc 149:A1184–A1189
Hu Y, Doeff MM, Kostecki R et al (2004) J Electrochem Soc 151:A1279–A1285
Chen JJ, Whittingham MS (2006) Electrochem Commun 8:855–859
Liang GC, Ou XQ, Wang L et al (2008) J Power Sources 184:538–542
Gao G, Liu AF, Hu ZH et al (2011) Rare Met 30:433–438
Lee J, Teja AS (2005) J Supercrit Fluids 35:83–90
Dokko K, Koizumi S, Sharaishi K et al (2007) J Power Sources 165:656–659
Dokko K, Shiraishi K, Kanamura K (2005) J Electrochem Soc 152:A2199–A2202
Meligrana G, Gerbaldi C, Tuel A et al (2006) J Power Sources 160:516–522
Liang GC, Wang L, Ou XQ et al (2008) J Power Sources 184:538–542
Shu HB, Wang XY, Wu Q et al (2012) J Electrochem Soc 159:A1904–A1911
Li XF, Zhang Z, Liu F et al (2013) Adv Mater Res 787:58–63
Zhang Z, Li XF, Hu YL et al. (2012) Chinese Patent CN102790214A
Chen M, Du CY, Song B et al (2013) J Power Sources 223:100–106
Shenouda AY, Liu HK (2009) J Alloys Compd 477:498–503
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, X., Zhang, X. & Zhang, Z. Synthesis of high-performance LiFePO4/C composite with a grape bunch structure through the hydrothermal method. Ionics 20, 1275–1283 (2014). https://doi.org/10.1007/s11581-014-1093-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-014-1093-4