
Abstract
LiNi1/3Co1/3Mn1/3O2 (LNMCO) powders were formed by a two-step synthesis including preparation of an oxalate precursor by “chimie douce” followed by a solid-state reaction with lithium hydroxide. The product was characterized by TG-DTA, X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared (FTIR), Raman spectroscopy, electron spin resonance (ESR), and SQUID magnetometry. XRD data revealed well-crystallized layered LNMCO with α-NaFeO2-type structure (R-3 m space group). Morphology studied by SEM and TEM shows submicronic particles of 400–800 nm with a tendency to agglomerate. The local structure investigated by vibrational spectroscopy (FTIR, Raman), ESR, and SQUID measurements confirms the well-crystallized lattice with a cation disorder of 2.6% Ni2+ ions in Li(3b) sites. Electrochemical tests were carried out in the potential range 2.5–4.5 V vs. lithium metal on samples heated at 900 °C for 12 h. Initial discharge capacity is 154 mAh/g at C/5, while a capacity of 82 mAh/g is still delivered at 10 C by the two-step synthesized LiNi1/3Co1/3Mn1/3O2 as cathode material.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the introduction of rechargeable lithium-ion batteries as power sources for portable electronic devices, several positive electrode materials have been extensively investigated as alternatives to LiCoO2, which presents instability at potential above 4.2 V. To power hybrid electric vehicles, plug-in hybrid electric vehicles, and electric vehicles, researchers are looking for new materials capable to deliver high power density, higher reversible capacity, good cyclability, and safety [1–3]. Among the candidates, several compounds with a layered structure have been suggested, including LiNi1−x CoO2 [4, 5] Li(Ni1−x Co x )1−y Al y O2 [6], LiNi1/2Mn1/2O2 [6–8], LiNi x Mn x Co1−2x O2 (0 ≥ x ≥ 1/3) and its derivatives [9–15]. Proposed by Ohzuku’s group, the particular composition LiNi1/3Co1/3Mn1/3O2 (LNMCO) [16, 17] exhibits much higher capacity, great structural stability, and enhanced safety [18].
Several synthetic methods are currently used to grow LNMCO powders such as solid-state reaction, sol–gel process, co-precipitation [19–21], microwave [22], self-propagating solid state [23], rheological phase [24], thermal polymerization [25], and microemulsion [26] techniques. In this respect, the most important factor is the selection of favorable precursor for the synthetic method to obtain single-phase and fine-sized powder. The wet-chemical co-precipitation method was adopted to prepare fine-sized precursor of nickel, cobalt, and manganese with desired particle morphology and homogeneous cationic distribution [27]. This method, which consists in the mixing of transition metal hydroxide precursor with hydrated lithium hydroxide, appears to be efficient because of the easy oxidation of elements during the precipitation reaction at the molecular level [28, 29].
From the electrochemical point of view, it has been demonstrated that the performance of LNMCO in high-power lithium-ion batteries is strongly dependent on the structural properties [30–34]. The main problem that still needs to be solved for such applications of LNMCO is the cation mixing between nickel and lithium-ions since the ionic radius of Ni2+ (0.69 Å) is close to that of Li+ (0.76 Å) for CN = 6 [35]. A partial occupation of Ni2+ by Li+ and Ni2+ generates a disorder in the structure that is damaging to the electrochemical properties [36, 37]. One aim of this work is to decrease this cation mixing by adjusting the synthetic conditions [38].
The goal of this work is to grow submicronic LiNi1/3Co1/3Mn1/3O2 powders with low cationic mixing to improve the rate capability of the cathode material. A two-step co-precipitation synthesis has been envisaged. The LNMCO powders were characterized using various physical methods to investigate the long- and short-range structure, including thermogravimetry–differential thermal analysis, X-ray diffraction (XRD), scanning electron (SEM) and transmission electron microscopy (TEM), elemental analysis, Fourier transform infrared (FTIR) and Raman scattering spectroscopy, superconducting quantum interference device (SQUID) magnetometry, and electron spin resonance (ESR). Finally, the electrochemical properties of LNMCO powders were tested in lithium cells using 1 M LiPF6 in EC/DEC solution as electrolyte.
Experimental
Sample preparation
The growth of LiNi1/3Co1/3Mn1/3O2 powders was ensured by a two-step process. First, the formation of the transition metal oxalate Ni1/3Co1/3Mn1/3C2O4·3H2O precursor was obtained by the co-precipitation method. Stoichiometric amounts of Ni(NO3)2·6H2O, Co(NO3)2·6H2O, and Mn(NO3)2·4H2O (cationic ratio of Ni/Co/Mn = 1:1:1) were dissolved in distilled water. Oxalic acid was used as a precipitator. The dissolved solution was added dropwise to 100 ml of 1 M oxalic acid with stirring until the complete precipitation of oxalate precursor. In order to keep the pH near neutral values (ca.7–8), an alkaline solution was timely added into the reaction vessel during co-precipitation period. The precipitated precursor was filtered and dried at 100 °C for 12 h. The second step consisted in the mixture of the oxalate precursor with stoichiometric amount of monohydrated LiOH followed by subsequent calcination at 900 °C for 12 h in air with intermittent grinding to obtain the final product. For comparison of electrochemical features, a reference LiNi1/3Co1/3Mn1/3O2 compound with similar porosity, surface chemistry, particle size, crystallinity, etc., but with a higher cationic disorder to justify the positive role of improved local environment, a sample was synthesized by the conventional wet-chemical method assisted by oxalic acid (single step).
Apparatus
Thermal analysis was carried out using a Perkin Elmer (TGA 7 series) gravimetric analyzer in the temperature range of 30–1,000 °C in air at a heating rate of 10 °C/min. The crystal structure was analyzed by XRD using a Philips X'Pert apparatus equipped with a CuKα X-ray source (λ = 1.5406 Å). XRD measurements were collected in the 2θ range 10–80° at low scanning rate. FTIR spectra were recorded with Bruker IFS 113 vacuum interferometer. In the far-infrared region (400–100 cm−1), the vacuum bench apparatus was equipped with a 3.5-μm-thick Mylar beam splitter, a globar source, and a DTGS/PE far-infrared detector. Raman spectra were measured using a Micro-Raman-Laser spectrometer model Lab-Ram (Horiba-Jobin-Yvon) microscope lens × 50, filter D2, diaphragm 400 μm, slit 150 μm. The spectra have been recorded with the red (632 nm) laser excitation.
Sample morphology was observed by SEM and TEM (JEOL, JEM-1230, Japan). The chemical compositions of the synthesized samples were confirmed by energy-dispersive X-ray spectroscopic analysis (EDAX) using an X-ray detector attached to the TEM instrument. Magnetic measurements (susceptibility and magnetization) were carried out with a fully automated magnetometer (MPMS-XL from Quantum Design) using an ultra-sensitive SQUID in the temperature range 4–300 K. A small quantity of sample was put into a small plastic vial, placed in a holder, and finally inserted into the helium drawer of the SQUID apparatus. The temperature dependence of the susceptibility data was recorded during heating of the sample using two modes—zero-field cooling (ZFC) and field cooling (FC)—to determine the magnetic behavior. The procedure is based on performing two consecutive magnetization measurements: in ZFC, the sample is first cooled down in the absence of magnetic field and then measured in an applied magnetic field increasing the temperature; the FC is performed in the same magnetic field, decreasing the temperature. For susceptibility measurements, constant magnetic field of 10 kOe was applied. Magnetic field in the range 0–30 kOe was applied for magnetization measurements.
Electrochemical tests were performed at room temperature with laboratory-scale cells which include a Li+ metal foil as a counter electrode. A positive electrode mixture was composed of 85 wt.% of active material, 10 wt.% of carbon black as a conductive agent, and 5 wt.% of polytetrafluoroethylene as a binder. The Li//LiNi1/3Co1/3Mn1/3O2 cells were housed in a Teflon laboratory-cell hardware employing a nonaqueous Li+ ion-conducting organic electrolyte 1 M LiPF6 in EC/DMC (1:1). A microporous polypropylene film (Celgard 2500) was used as a separator. The apparent lithium content of discharge compounds was estimated using the current passed and the mass of the electrode material. Different C-rate discharge currents were used in the potential range 2.5–4.5 V vs. Li0/Li+, and the data were recorded using a Mac-pile battery cycler.
Results and discussion
Thermal analysis
Figure 1a, b shows, respectively, the thermal gravimetry and differential scanning calorimetry (TG–DSC) curves of the formation of the precursor of transition metals oxalate Mn1/3Ni1/3Co1/3]C2O4 and the growth of the final product LiNi1/3Mn1/3Co1/3O2. In the TG–DSC graph of the oxalate precipitate, i.e., the sediment after the co-precipitation reaction, the profile has three steps of weight loss. The first two steps starts above 100 °C that shows a total of ca. 20 wt.% weight loss with two small broad endothermic peaks at 105 and 215 °C, which are attributed to the dehydration of the metal oxalate. The process for the formation of the oxalate is given by

TG–DSC curves at the heating rate of 10 °C/min in air a of oxalate precursor [Mn1/3Ni1/3Co1/3]C2O4·3H2O and b of the LiOH·H2O:[Mn1/3Ni1/3Co1/3]C2O4·3H2O mixture
We obtained violet-colored sediment with pH at ca. 8. Based on the color of the sediment, the oxalate precursor [Mn1/3Ni1/3Co1/3]C2O4· has a monoclinic structure [39]. Then, upon increasing the temperature up to 300 °C, anhydrous [Mn1/3Ni1/3Co1/3]C2O4 is decomposed into metal oxide and carbon dioxide gas.
Figure 1b shows the TG–DSC curves recorded during the growth of LiNi1/3Mn1/3Co1/3O2 prepared from the mixing of [Mn1/3Ni1/3Co1/3]C2O4· with LiOH·H2O by 1:1 molar ratio. A small endothermic peak was observed at ca. 200 °C due to the dehydration of the lithium hydroxide component. There is one strong exothermic peak located at ca. 505 °C attributed to the decomposition of the oxalate precursor and the beginning of crystallization of LiNi1/3Mn1/3Co1/3O2 powders according to the reaction
Actually, the use of the two-step co-precipitation method associates advantages of a low-temperature route with the preparation of the metal oxalate di-hydrated precursor (Ni1/3Co1/3Mn1/3)C2O4 of the monoclinic structure, which crystallizes into (Ni1/3Co1/3Mn1/3)3O4 by thermal treatment in air. The results of this method are a low defective structure and the formation of spherical-like particles with a narrow size distribution as shown in the next section.
Structure and morphology
Figure 2 displays the XRD spectrum of LiNi1/3Mn1/3Co1/3O2 synthesized with calcination at 900 °C for 12 h in air. The diffraction pattern shows well-defined Bragg lines that correspond to the rhombohedral α-NaFeO2 structure (R-3 m space group). As expected, the two dominant peaks correspond to the (003) and (104) lines with an intensity ratio I (003)/I (104) = 1.67. The splitting of the lines (006)/(012) and (018)/(110) reveals the highly ordered layered structure of the LNMCO powders [4]. This is also a good indication for small cation mixing. An accurate evaluation of the cation mixing, the amount of Ni2+(3b), will be made in the following by magnetic measurements as these are powerful tools when 3d transition metal are involved.

XRD pattern of LiNi1/3Mn1/3Co1/3O2 prepared via oxalate co-precipitation method
The lattice parameters of LiNi1/3Mn1/3Co1/3O2 were calculated by a least squares method using ten diffraction peaks in the hexagonal sitting as a = 2.867(4) Å, c = 14.246(7) Å, V = 100.84(8) Å3, and c/a = 4.978. These values are in good agreement with those reported by Ohzuku and Makimura [11, 12]. The coherent length L = 55 nm calculated from the Scherrer formula shows that each particle contains several mono-crystallites. Note that the c/a ratio corresponding to lower rhombohedral distortion is also a good indication for the cationic disorder in LNMCO materials for which the local symmetry is lowered due to the presence of Ni2+(3b).
Figure 3 shows the SEM image (a) and the TEM picture (b) for the LNMCO powders heat-treated at 900 °C. Well-developed particles with regular shape, almost spherical, are observed, indicating good crystallinity of the synthesized powders. The well-defined particles were attributed to the careful synthesis of the oxide, including extensive grinding between successive calcinations. The TEM image clearly shows similar results. The particles have submicron size in the range 400–600 nm. Their shape tends to be spherical with a narrow grain size distribution.

SEM image (a) and TEM picture (b) of the LNMCO powders heat-treated at 900 °C for 12 h in air
Figure 4 shows the results of the elemental analysis carried out by EDAX using an X-ray detector attached to the TEM instrument. This spectrum reveals the presence of Co, Ni, Mn, and oxygen as expected for stoichiometric LiNi1\3Mn1\3Co1\3O2 material. In the EDS spectrum, other peaks found related to Cu are only due to the Cu micro-grid sample holder [40].

EDAX spectrum of LiNi1/3Mn1/3Co1/3O2 prepared via oxalate co-precipitation method with calcination at 900 °C for 12 h in air
Vibrational spectroscopy
Because Raman and FTIR spectroscopy are sensitive to the short-range environment of oxygen coordination around cations in oxide lattices, the frequencies and the relative intensities of the bands are the fingerprints of the coordination geometry and the oxidation state of the cations. Figure 5 displays the vibrational features of LiNi1\3Co1\3Mn1\3O2: (a) the FTIR absorption spectrum and (b) the Raman scattering spectrum. Considering the space group R-3 m for LiNi1\3Co1\3Mn1\3O2 crystal that corresponds to the D 5 3d spectroscopic symmetry, the group factor theory provides the vibrational active modes represented as follows:
in which A 1g and E g are Raman-active and the remaining A 2u and E u are infrared active modes [41]. The A 1g and A 2u modes originate from the M–O symmetrical stretching, while E g and E u come from the O–M–O bending vibrations [5]. Because of the D 3d site group of the Wyckoff (3b) for the lithium ions, the Li cage mode is only active in infrared. Since the compound has three M-ions, we expect 3A 1g modes and 3E g modes that overlap to give rise to the two broad A 1g and E g structures in Fig. 5a. The best fit to the Raman spectrum has then been achieved, starting from a set of three individual bands of Lorentzian shape for the overlapping A 1g band profile and same for the E g band profile (see Fig. 5b). The results are given in Table 1. The E g bands are centered at 475, 482, and 516 cm−1 and the A 1g bands are centered at 576, 602, and 639 cm−1 for M = Ni, Co, and Mn, respectively. These positions compare well with the corresponding bands of LiNiO2, LiCoO2, and λ-LiMn2O4, respectively [42]. Note also that the width of the υ 1 bending vibrations of Ni–O–Ni is larger than for other cations, which gives evidence of the degree of Ni cationic disorder in this sample, in agreement with the structural analysis in the following section.

The FTIR absorption spectrum LiNi1\3Co1\3Mn1\3O2 is presented in Fig. 5a. According to factor group analysis, the IR absorption bands are situated in three distinct regions. The lower-wavenumber band at 246 cm−1 is attributed to the Li cage mode. This band vanishes with Li extraction from the LNMCO framework. The medium-wavenumber region 400–520 cm−1 corresponds to the O–M–O asymmetric bending modes, while at above 520 cm−1 the IR modes are due to the asymmetric stretching modes of the MO6 octahedra. The best fit to the IR spectrum has then been achieved, starting from a set of three individual bands of mixed Gaussian–Lorentzian shape. It is reported in Fig. 5a, and the results are given in Table 2.
Magnetic and electronic properties
The temperature dependence of the reciprocal magnetic susceptibility χ −1m (T) = H/M and the isothermal magnetization curves M(H) of LNMCO samples prepared from oxalate co-precipitation are shown in Fig. 6a, b respectively. Measurements were carried out in the ZFC mode with H = 10 kOe. At T > 80 K, the quasi-linear variations of χ −1m (T) can be described by Curie–Weiss law:
with Θ p as the Curie–Weiss temperature, C p as the Curie constant related to the effective magnetic moment μ eff, N as the number of metal ions in 1 mol of product, and k B as the Boltzmann constant. The values of the fitting parameters are C p = 0.93 emu/mol, μ eff = 2.74, μ B, and Θ p = −74 K, which indicate the dominant antiferromagnetic interactions. Taking into account that the magnetic moments carried by Ni2+ and Mn4+ are 2.83 μ B and 3.87 μ B, respectively, while Co3+ is not magnetic [43, 44], the theoretical value of μ eff in the absence of Ni2+(3b) defects would be:

a The temperature dependence of the reciprocal magnetic susceptibility χ −1m (T) = H/M and b the isothermal magnetization curve M(H) of LNMCO samples prepared from oxalate co-precipitation
The experimental value of μ eff is close to this theoretical value. In Fig. 6b, the magnetization curves show a non-linearity for T < 50 K. Following our prior works [21], this feature is attributed to the presence of Ni2+(3b) defects that form Mn4+(3a)–Ni2+(3b) ferromagnetic pairs (FMP) at low temperature according to the Goodenough–Kanehori rules [45]. The contribution of FMP to the curves M(H) gives rise to an enhancement of the magnetization responsible for the decrease of χ −1m (T < 50 K) as shown in Fig. 6a.
At 4 K, the magnetization associated with FMP is approximately saturated under the application of an external magnetic field (H = 30 kOe). We can then estimate the amount of Ni2+(3b) defects as the ratio between the magnetic moment at saturation M s of the ferromagnetic component of the magnetization and the moment (per chemical formula) at saturation that the sample would have if all the Mn and Ni ions are fully spin-polarized such as
With the M s = 246 emu/mol, the magnetic moment at saturation estimated from the linear extrapolation to H → 0 of M(H) at T = 4.2 K taken in the high magnetic field range, the amount of Ni2+ at 3b sites is 2.6%. This value can be compared with the cationic disorder of a LiNi1/3Mn1/3Co1/3O2 sample synthesized using the conventional wet-chemistry assisted by oxalic acid. For this material, the cationic mixing is evaluated to be 3.2% compared with 2.6% for the present study. Thus, it is evident that the synthetic method plays an important role in the crystal chemistry of LNMCOs.
Figure 7 shows the ESR spectrum recorded at 300 K for the LiNi1/3Mn1/3Co1/3O2 calcined at 900 °C for 12 h. The ESR feature consists of a broad signal with peak-to-peak width ∆H pp ≈ 1,200 G, which agrees with previous data of the same structure [43]. The ESR signal centered close to 3,200 G is the position expected for uncorrelated spins with the gyromagnetic factor g = 2.033 for the X-band microwave frequency we have used. The LiNi1/3Mn1/3Co1/3O2 oxide contains two magnetic ions, Ni2+ and Mn4+, and one Co3+ nonmagnetic ion. Because of its even number of d-electrons, Ni2+ is a non-Kramers ion and the ESR spectrum is very sensitive to the interactions with the environment. Consequently, the ESR signal of Ni2+ can then be detected only when this ion is present as a residual impurity [46]. The paramagnetic resonance detected in Fig. 7 is then attributable to the presence of Mn4+ ions that carry a half-integer spin (S = 3/2) and are then ESR-active. The effective g-factor for the Mn4+ ions close to g = 2.0 is characteristic of the octahedral environment [47, 48]. When the crystal field symmetry is lower, for instance, a smaller g-value is observed, like g = 1.94 in MnO2 for instance [49]. This is consistent with our structural analysis that has evidenced the good crystallinity of the sample.

Electron spin resonance spectrum of LiNi1/3Mn1/3Co1/3O2 prepared via oxalate co-precipitation method at room temperature. The magnetic field was scanned in the range 0–10 kG
Electrochemical tests
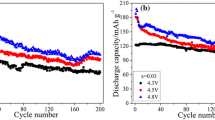
Figure 8 describes the first charge–discharge profile of a Li//LiNi1/3Mn1/3Co1/3O2 cell. The electrochemical tests were performed at 0.2 C-rate between 2.5 and 4.5 V vs. Li0/Li+ with 1 M LiPF6 in EC/DEC (1:1) as electrolyte. The curves shown in Fig. 8 are typical of a well-crystallized LNMO material without existence of potential plateau. After a small amount of Li has been extracted from the layered structure, the cell potential increases rapidly from the OCV 3.15 V to 3.65 V and then increases smoothly up to 4.5 V, corresponding to the extraction of 0.6 Li, which means that almost all Ni2+ ions were ionized to Ni4+. This cell delivers an initial discharge capacity 152 mAh/g; thus, 12 mAh/g retention (irreversible) capacity was observed, giving 93% discharge–charge efficiency. In the same potential window, the electrochemical features of Li//LiNi1/3Mn1/3Co1/3O2 cells have been investigated as a function of C-rate since the rate capability is one of the important battery performances. Figure 9 shows the discharge profile for various C-rates in the range from C/5 to 10 C. Slight polarization was observed even at rather high 2 C-rate. For this range of density of current, the discharge capacity values measured for 36 continuous cycles show a decay of 0.35% per cycle. Thus, there is an about 12.5% capacity loss at the end of the 36 cycles.

Initial charge–discharge curve of LiNi1/3Mn1/3Co1/3O2 at 0.2 C in LiPF6 electrolyte between 2.5 and 4.5 V vs. Li/Li+

Discharge curves of LiNi1/3Mn1/3Co1/3O2 at various C-rates in LiPF6 electrolyte between 2.5 and 4.5 V vs. Li/Li+
Figure 10 displays the Ragone plot for the Li//LiNi1/3Mn1/3Co1/3O2 cells investigated after 5 cycles in the same conditions. We compared cells using two LiNi1/3Mn1/3Co1/3O2 cathode materials synthesized by different routes: (a) the conventional wet-chemistry assisted by oxalic acid, which provides material with 3.2% cationic disorder, and (b) the two-step oxalate method for which Ni2+(3b) = 2.6%. It is obvious that the lower cationic disorder delivered the high capacity retention. Even at high C-rate, the reversible capacity was 83 mAh/g, which is 50% of the initial discharge capacity.

Ragone plots of the Li/ LiNi1/3Mn1/3Co1/3O2 cells using 1 M LiPF6 in EC-DEC electrolyte. Cells were cycled between 2.5 and 4.5 V vs. Li0/Li+. Cathode materials were synthesized by the conventional wet chemical method assisted by the oxalic acid (a) and the two-step oxalate route (b)
Note that LiNi1/3Mn1/3Co1/3O2 is a very well-studied compound whose electrochemical properties have been reported to be affected by precursors, synthesis routes, and a lot of factors. This work justifies the positive role of the preparation method in reducing cationic disorder and, as a result, electrochemical properties are also improved (especially at high rates); in the case of transition metal oxides with a layered framework, the structural defects play an important role in the electrochemical performance of these positive electrode materials. It is recognized worldwide that structural defects such as cationic disorder are attributable to a decrease of lithium-ion transport in the cathode material. The comparison between different LiNi1/3Mn1/3Co1/3O2 samples is meaningful, and the difference between the electrochemical properties is attributable to the only parameter that has varied in the process, namely, the cation mixing.
Conclusion
In this work, we have studied the local structure, morphology, and magnetic and electrochemical properties of LiNi1/3Co1/3Mn1/3O2 cathode material prepared by oxalate co-precipitation. Thermal analysis showed that LiNi1/3Co1/3Mn1/3O2 started to crystallize at around 600 °C. Pure-phase LiNi1/3Co1/3Mn1/3O2 was obtained when powders were heat-treated at 900 °C for 12 h in air, showing submicron-sized particles at 400–600 nm and spherical in shape. The local structure was characterized by FTIR and Raman spectroscopy emphasized the layered structure of LiNi1/3Co1/3Mn1/3O2. From an analysis of magnetic measurements, the degree of cation mixing was estimated to be Ni2+(3b) = 2.6%. High reversible capacity and good rate capability were observed from testing this oxide as cathode in lithium batteries with 1 M LiPF6 in EC-DEC as electrolyte.
References
Dahn JR, Zheng T, Thomas CL (1997) J Electrochem Soc 145:851
Zaghib K, Ravet N, Gauthier M, Gendron F, Mauger A, Goodenough JB, Julien CM (2006) J Power Sources 163:560
Whittingham MS (2004) Chem Rev 104:4271
Rougier A, Gravereau P, Delmas C (1996) J Electrochem Soc 143:1168
Julien C, Letranchant C, Rangan S, Lemal M, Ziolkiewicz S, Castro-Garia S, El-Farh L, Benkadour M (2000) Mater Sci Eng B 76:145
Venkatraman S, Manthiram A (2005) Solid State Ionics 176:291
Ohzuku T, Makimura Y (2001) Chem Lette 744
Kang KS, Meng YS, Breger J, Grey CP, Ceder G (2006) Science 311:977
MacNeil DD, Lu Z, Dahn JR (2002) J Electrochem Soc 149:A1332
Lu Z, MacNeil DD, Dahn JR (2001) Electrochem Solid State Lett 4:A200
Ohzuku T, Makimura Y (2001) Chem Lett 1:642
Yabuuchi N, Ohzuku T (2003) J Power Sources 119:171
Cho J, Manthiram A (2005) J Electrochem Soc 152:A1714
Hwang BJ, Tsai YW, Cariler D, Ceder G (2003) Chem Mater 15:376
Tsai YW, Hwang BJ, Ceder G, Sheu HS, Liu DG, Lee JF (2005) Chem Mater 17:3191
Kim JM, Chung HT (2004) Electrochim Acta 49:937
Koyama Y, Tanaka I, Ohuzuku T (2003) J Power Sources 119–121:644
Guo J, Jiao L, Yuan H, Li H, Zhang M, Wang Y (2006) Electrochim Acta 51:3731
Li D, Muta T, Zhang L, Yoshio M, Noguchi H (2004) J Power Sources 132:150
Hu SK, Cheng GH, Cheng MY, Hwang BJ, Santhanam R (2009) J Power Sources 188:564
Park BC, Bang HJ, Yoon CS, Myung ST, Prakash J, Sun YK (2007) J Electrochem Soc 154:A520
Shen BJ, Ma JS, Wu HC, Lu CH (2008) Mater Lett 62:4075
He YS, Ma ZF, Lia XZ, Jiang Y (2007) J Power Sources 163:1053
Ren H, Wang Y, Li D, Ren L, Peng Z, Zhou Y (2008) J Power Sources 178:439
Ding CX, Meng QS, Wang L, Chen CH (2009) Mater Res Bull 44:492
Lu CH, Lin YK (2009) J Power Sources 189:40
Chuan-Fu Z, Ping Y, Xi D, Xuan X, Jing Z, Z Liang Y (2009) Trans Nonferrous Met Soc China 19:635
Mijung N, Lee Y, Cho J (2006) J Electrochem Soc 153:935
Choi J, Manthiram A (2005) Electrochem Solid State Lett 8:C102
Sinha NN, Munichandraiah N (2010) J Electrochem Soc 157:A647
Konishi H, Yuasa T, Yoshikawa M (2011) J Power Sources 196:6884
Gao J, Kim J, Manthiram A (2009) Electrochem Commun 11:84
Liu H, Tan L (2011) Mater Chem Phys 129:729
Zhang X, Jiang WJ, Zhu XP, Mauger A, Lu Q, Julien CM (2011) J Power Sources 196:5102
Shannon RD (1976) Crystallogr Acta A32:751
Lee K-S, Myung S-T, Prakash J, Yashiro H, Sun Y-K (2008) Electrochim Acta 53:3065
Shaju KM, Bruce PG (2007) J Power Sources 174:1201
Zhang X, Mauger A, Lu Q, Groult H, Perrigaud L, Gendron F, Julien CM (2010) Electrochim Acta 55:6440
Cho TH, Shiosaki Y, Noguchi H (2006) J Power Sources 159:1322
Abuzeid HM, Hashem AM, Narayanan N, Ehrenberg H, Julien CM (2011) Solid State Ionics 182:108
Julien C, Massot M (2002) Solid State Ionics 148:53
Julien C (2000) Solid State Ionics 136:887
Abdel-Ghany A, Mauger A, Gendron F, Zaghib K, Julien CM (2007) ECS Trans 3(36):137
Zhang X, Jiang WJ, Mauger A, Lu Q, Gendron F, Julien CM (2010) J Power Sources 195:1292
Goodenough JB (1963) Magnetism and the chemical bond. Wiley, New York
Alonso PJ, Alcala R, Spaeth JM (1990) Phys Rev B 41:10902
Okubo MS, Elmasry F, Zhang W, Fujisawa M, Sakurai T, Ohta H, Azuma M, Sumimova OA, Kumada N (2010) J Phys Conf Ser 200:022042
Andresen HG (1961) J Chem Phys 35:1090
Kakazey M, Ivanova N, Boldurev Y, Ivanov S, Sokolsky G, Gonzalez-Rodriguez JG, Vlasova M (2003) J Power Sources 114:170
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hashem, A.M., El-Taweel, R.S., Abuzeid, H.M. et al. Structural and electrochemical properties of LiNi1/3Co1/3Mn1/3O2 material prepared by a two-step synthesis via oxalate precursor. Ionics 18, 1–9 (2012). https://doi.org/10.1007/s11581-011-0635-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-011-0635-2