
Abstract
A Ramularia-like hyphomycete was discovered on uredinia of Phakopsora ampelopsidis on leaves of wild Ampelopsis brevipedunculata and cultivated Parthenocissus tricuspidata in several cities in Taiwan. The micromorphology of this fungicolous fungus is similar to that of species of the genus Ramularia which are mostly plant parasites but some species grow on rust fungi. Analyses of SSU and LSU rDNA and RPB2 gene sequence data and of ultrastructural features observed by scanning and transmission electron microscopy revealed that the fungus represents a novel lineage within the Ustilaginomycotina. The name Quasiramularia phakopsoricola is proposed for this new species in a new genus, a new family, and a new order. Sequence data obtained for ITS1, 5.8S, and ITS2 rDNA, however, did not match to any known fungal lineage. Bioinformatics analyses showed that these sequence data are extremely GC poor, but most probably do not represent a pseudogene. Extreme deviations of GC content can be observed in several lineages of Basidiomycota. Such variations affect the results of phylogenetic analyses and are an important source of artifacts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ramularia is a genus including species of Ascomycota that are mainly plant parasites, but some species typically colonize other leaf-inhabiting fungi, mainly rust fungi (Pucciniales/Uredinales) (Braun 1998). Plant-associated Ramularia-like anamorphs of species of Ustilaginomycotina (Basidiomycota) were sometimes confused with Ramularia spp. in the past (Braun 1995). Mostly hyaline conidiophores and conidia formed by sympodial conidiogenesis are features common to Ramularia spp. in the Ascomycota and to species within the Basidiomycota belonging to Doassansia (anamorph Savulescuella), Doassansiopsis (anamorph Doassansiella), Entyloma (anamorph Entylomella), Exobasidium (dimorphic), Quambalaria (exclusively anamorphic), Urocystis (anamorph Paepalopsis) (Braun 1995; Simpson 2000; Aime et al. 2018), and to the recently discovered genus Capitulocladosporium (exclusively anamorphic) with unknown habitat (Sun et al. 2018).
On living leaves of several plants of Vitaceae infected with rust fungi, we detected a Ramularia-like fungus which could not be identified as a species of Ramularia or of other similar genera. The 5.8S rDNA of the ITS region suggested a relationship with Basidiomycota, but the flanking regions did not match in BLAST searches and indicated a non-typical DNA region, with extremely low guanine-cytosine (GC) content. This fungus and its rDNA regions were, therefore, studied in detail. In particular, we considered the possible presence of a pseudogene and we investigated the impact of GC-poor rDNA sequences on results of phylogenetic analyses in Basidiomycota.
A pseudogene is a non-functional rDNA copy. It exhibits higher variability in conserved regions, it is GC poor, and its secondary structure is not as stable as in functional copies. Such deviating rDNA copies are known to be present in fungal genomes (Stadler et al. 2020) and can produce artifacts in phylogenetic analyses, if treated as a standard rDNA copy. Therefore, the structure of the rDNA from the Ramularia-like fungus was investigated in order to distinguish functional from potentially non-functional rDNA. GC content itself differs between species and between different DNA regions in the same chromosome (Testa et al. 2016). GC content heterogeneity between taxa is responsible for reconstruction biases such as contradicting topologies and poor node resolution across the whole tree of life, including fungi (Collins et al. 2005; Romiguier and Roux 2017). The reasons underlying such strong biases are not well known, but the substitution rate, which differs between GC-rich and GC-poor regions, is important in this context (Romiguier and Roux 2017; Wang et al. 2017). The phenomenon of the GC content bias in phylogenetic analyses of fungi is studied here for the first time, and our study provides a first comparison of GC content in rDNA sequences across all major lineages of Basidiomycota.
Materials and methods
Collection and isolation of strains
Leaves of wild Ampelopsis brevipedunculata (Maxim.) Trautv. and cultivated Parthenocissus tricuspidata (Siebold & Zucc.) Planch. (Vitaceae) infected with a rust fungus overgrown by a whitish hyphomycete were collected at way- and roadsides in three cities, namely Taipei City, New Taipei City, and Taoyuan City, in northern Taiwan. Air-dried specimens were deposited in the fungal collection of the National Museum of Natural Science (TNM), Taichung, Taiwan.
Conidia observed under a dissecting microscope were picked up with a flamed acupuncture needle and transferred to Petri dishes containing corn meal agar (Fluka) with 0.2% chloramphenicol. Isolates were grown at diffuse daylight and room temperature. Representative strains were deposited at the Bioresource Collection and Research Centre (BCRC), Hsinchu, Taiwan.
Light microscopy
For light microscopical preparations, cells of the uredinicolous fungus were removed using tweezers or tape, and mounted on a slide. Most measurements and drawings of the micromorphological characteristics were made in 10% KOH mounts under the light microscope with 1000 × magnification. Measurements were made from n = 30 replicates and were presented as mean value ± standard deviation and extreme values in brackets. The scale bar of 3 cm in the drawings was equal to 10 µm in the microscope. The drawings were made with a pencil, then depicted with technical pens with tracing paper. All illustrations were composed and modified with Photoshop CS6.
Scanning electron microscopy (SEM)
Pieces of leaves with overgrown rust sori (WIC009) were fixed by immersion in 2% (w/v) aqueous osmium tetroxide (OsO4) for 12 h at 4 °C in the dark, washed three times in distilled water (15 min each wash) to remove the excess osmium tetroxide and then dehydrated in an ethanol series each for 15 min and steps from 10 to 90% ethanol. After washing the material in 95% ethanol, the material was kept in absolute ethanol for three periods of 15 min, with fresh ethanol each time. Ethanol was then replaced with acetone in 2:1 and 1:2 (ethanol:acetone) steps followed by three changes with absolute acetone (15 min each). The material was transferred to the pressure chamber of a Hitachi HCP-2 critical point dryer. Liquid CO2 was used as the drying agent. Following replacement of the acetone with liquid CO2, the material was maintained within the pressure chamber for an hour at 20 °C and then the acetone-contaminated liquid CO2 replaced with fresh liquid CO2. Slow heating of the liquid CO2 to 5° above its critical point (31.1 °C) followed by the gradual release of pressure at the same temperature (ca. 37 °C) resulted in dehydrated specimens in a gaseous phase at atmosphere pressure. Critical point dried material was mounted on aluminum stubs with double-sided conductive adhesive tape or carbon cement and left overnight in a desiccator for the cement to dry. Mounted material was coated with gold (40–50-nm thickness) in a Hitachi E-1045 Ion Sputter Coater and was examined in a Hitachi S4700 Field Emission Scanning Electron Microscope at 3 kV. Unpublished photographs of the same specimen of R. pusilla Unger as in Kirschner (2009) was included for comparison.
Transmission electron microscopy (TEM)
Material from a culture and from leaves (WIC009 and R. Kirschner 4383) was fixed in a 4% (V/V) glutaraldehyde solution buffered with 0.1 M sodium cacodylate (pH 7.2) containing 0.25 M sucrose, for 4 h at room temperature. Sucrose was added in order to adjust the osmolality of the fixative to that of seawater. Excess glutaraldehyde was removed by washing successively in 3 changes (15 min each change) of 0.1 M sodium cacodylate buffer (pH 7.2) containing a decreasing concentration of sucrose (i.e., 0.225, 0.125, 0.0625 M) to sucrose-free buffer. Material was post-fixed in 2% (W/V) osmium tetroxide in 0.1 M sodium cacodylate buffer (pH 7.2) for 12 h at 4 °C in the dark, then washed with 0.1 M sodium cacodylate buffer for 30 min with 2 changes to remove excess osmium tetroxide followed by distilled water. Fixed and washed material was dehydrated to acetone as described for SEM. Dehydrated material was embedded in Agar 100 epoxy resin (Spurr 1969), dehydrated to absolute acetone and slowly infiltrated with resin by transfer to 1:3, 1:1, 3:1 resin:acetone mixtures and finally to pure resin (araldite, Ciba Geigy, Cambridge) and polymerized in an oven. Material was maintained in each resin and placed in a desiccator for at least 2 h at room temperature and then transferred to a 70 °C for 3 days to complete polymerization. Under low magnification, resin was trimmed away from embedded material with a razor blade to form a trapezoid block. Ultrathin sections were made, treated, and examined in a Philips CM 12 at 80 kV as in Kirschner (2009). Additional sections were cut from the resin blocks with a Reichert Jung Ultracut E ultramicrotome using glass knives, floated on water, and were picked up on coated copper grids PELCO® Center Marked Grids, 75 mesh, 3.0 mm. Staining with drops of centrifuged lead citrate for 15 to 30 min (Reynolds 1963) was followed by washing and subsequent post-staining with centrifuged, saturated uranyl acetate in 50% ethanol for 30–60 min in the dark. Material on grids was examined in a JEOL 100S TEM at 80 kV or a Hitachi H 600 TEM at 75 kV.
Molecular analysis
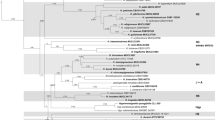
DNA extraction, PCR, and sequencing were largely performed as in Wei and Kirschner (2017). For amplification of the ribosomal RNA gene region, primers ITS1F and ITS4 (Gardes and Bruns 1993; White et al. 1990) were used for amplifying the internal transcribed spacer (ITS, including ITS1, 5.8S rDNA, ITS2, and fragments of the small and large ribosomal subunit RNA genes), primers NL1 and NL4 (Kurtzman and Robnett 1997) for the partial nuclear ribosomal large subunit gene (LSU), and primers NS3 and NS4 for the partial nuclear ribosomal small subunit gene (SSU) (White et al. 1990). For the DNA-directed RNA polymerase II second largest subunit gene (RPB2), primers RPB2-5F and PRB2-7R were applied (Liu et al. 1999). The thermocycler programming followed the literature cited for the primers. PCR products were sequenced by Mission Biotech (Nankang District, Taipei City) with the same primers used for the PCR. The two single read ABI files of each sequenced PCR product were edited pairwise with CodonCode Aligner version 4.0.1 (CodonCode Corporation, USA). The sequences were submitted to BLAST searches at GenBank (https://blast.ncbi.nlm.nih.gov/) for primary testing of the technical sequence quality and matches with highly identical sequences. The sequences were deposited in GenBank and the DNA Data Bank of Japan. The datasets of LSU (61 sequences), SSU (59), and RPB2 (51), sequences were assembled based on the most similar sequences from GenBank and by adopting the taxon sampling from Sun et al. (2018) and McTaggart et al. (2020) with some modifications (ESM4). From the RPB2 gene sequence data, the intron sequences were removed, exons translated to amino acids, and further analyzed as a protein sequence. For the LSU and SSU regions, the members of Moniliellales were hardly alignable, formed a long branch in the phylogenetic analyses (ESM4) and were omitted from further rDNA-based analyses. Sequence alignment was done in MAFFT ver. 7.222 (Katoh and Standley 2013) under the Q-INS-i strategy (considering rRNA secondary structure) for LSU and SSU and G-INS-i strategy for RPB2. In rDNA data, both gaps and variable regions were removed using Gblocks v. 0.91b (Talavera and Castresana 2007) with less stringent selection allowing smaller final blocks and gap positions within the final blocks. Single gene and concatenated datasets (LSU + SSU, LSU + SSU + RPB2) were analyzed. Bayesian searches (MB) were conducted with MrBayes 3.0 (Ronquist and Huelsenbeck 2003) and 10 million replicates estimated together with burn-in value in Tracer v1.5 (Rambaut et al. 2018). The best nucleotide substitution model for each partition was determined in jModelTest v. 2.1.1 (Darriba et al. 2012). Maximum likelihood (ML) searches were conducted in IQ-TREE v. 2.1.3 (Minh et al. 2020) with automated model selection for each partition using ModelFinder (Kalyaanamoorthy et al. 2017) and various settings such as FreeRate (+ R) model and General Heterogeneous evolution On a Single Topology (GHOST) model of sequence evolution (+ FO*H4) which naturally accounts for heterotachous evolution. Branch supports were assessed by the standard nonparametric bootstrap approximation (“-b 1000 “). The phylogenetic analyses were carried out with the (-p option) that allowed each partition to have its own evolutionary rate. Entorrhiza parvula Vánky identified by Riess et al. (2019) was chosen as outgroup because it is a taxon located at the base of Basidiomycota. Detailed dataset description and selected models are presented in Fig. 1. The presence of repeat-induced point mutation (RIP) in ITS sequences of four Quasiramularia strains was determined using Ripcal2 software (Hane and Oliver 2008).

Phylograms obtained from a maximum likelihood search in IQTree v. 2.1.3 showing the placement of Quasiramularia phakopsoricola. The trees were constructed using LSU and SSU rDNA nucleotide (left) and RPB2 amino acid sequences (right). Dataset descriptions and models are presented for each tree. Bootstrap support (BS) and Bayesian posterior (PP) probabilities are given at the nodes. The tree is rooted with Entorrhiza parvula. The scale bar indicates expected changes per site. Dashes (–) indicate support less than 70% BS or 0.7 PP for a particular clade. Asterisks (*) indicate mismatches of the topology between trees generated by IQTree and MrBayes
The GC content of ITS1, ITS2, 5.8S, LSU (28S), and SSU rDNA sequences of Quasiramularia phakopsoricola was compared to the GC content in sequence data deposited in the NCBI GenBank database. Therefore, we downloaded rDNA sequences from Basidiomycota and Entorrhizomycetes (25.12.2020, search based on the title, length restriction 150–4000 bp) and selected sequentially unique entries. As a result, we obtained 133,463 sequences. ITSx extractor (Bengtsson‐Palme et al. 2013) was used to remove chimeric sequences and to extract full-length ITS1 (31,143 sequences), ITS2 (50,659), 5.8S (93,890), and partial LSU sequences (56,382). For LSU sequences, only those longer that 300 bp were used for further analyses (8153 sequences). In addition, ITSx extracted entries without detectable ITS region motives (28,046 sequences), which belonged to sequences of solitary SSU (5857 sequences) and LSU sequences longer than 300 bp (23,490). They were separated from each other based on the title description. Datasets of LSU regions extracted from ITS-LSU entries and solitary LSU sequences were merged for further analyses. For correct GC content analysis across multiple fungal lineages, it is necessary to compare homologous positions of comparable length only. Therefore, SSU and LSU datasets were aligned in MAFFT ver. 7.222 (Katoh and Standley 2013). Hardly alignable sequences from species of the order Cantharellales and sequences with introns or strings of ambiguous nucleotide positions (coded as N) longer than 30 bp were removed. The reliability of the alignment was checked visually and using maximum likelihood trees generated in PhyML 3.0 (Guindon et al. 2010) using the SH-like support method suitable for large datasets.
In the LSU dataset, sequences were trimmed to the D1 variable domain (positions from 200 to 520 in Schizophyllum commune Fr. MT463493), because of the high length coverage across taxa. Finally, sequences shorter than 280 bp were removed. The final LSU dataset contained 30,734 sequences with a mean length of 317 bp, SD = 5.8, and a maximum length of 367 bp.
The SSU alignment was divided into two parts, roughly corresponding to regions before and behind the bp position 550 of S. commune KT715706. This step allows to compare GenBank entries of different lengths. Sequences shorter than 499 bp (first part) and 400 bp (second part) were removed from each dataset. The first dataset contained 5038 sequences (mean 530, SD = 17.6, maximum 562 bp) and the second dataset 5368 sequences (mean 435, SD = 3.3, maximum 467 bp). The dataset of full-length 5.8S rDNA sequences contained 90,621 entries (minimum length 154, mean 158.2, SD = 2.6, maximum 219), the dataset of full-length ITS2 rDNA sequences 50,590 entries (minimum length 67, mean 216.4, SD = 1.7, maximum 978 bp), and the dataset of full-length ITS1 rDNA sequences 31,083 entries (minimum length 65, mean 211, SD = 46.3, maximum 1181 bp).
Finally, all datasets were divided into six groups according to systematic relationships (Agaricomycotina, Entorrhizomycetes, Pucciniomycotina, Quasiramularia, Ustilaginomycotina, and Wallemiomycotina). The GC content in sequences of different loci was calculated in DaMBE ver 6.4.42 (Xia and Xie 2001). All sequence manipulations were done in Seed2 (Větrovský et al. 2018). Datasets are presented in Electronic Supplementary Materials 2 and 5.
The secondary structures of the ITS2 sequences and associated minimum free energy were predicted using the ITS2 Database II (Selig et al. 2008) and RNAFOLD server (Hofacker et al. 1994). The 5.8S secondary structure was predicted in the RNAFOLD server. The 5.8S secondary structure was inspected for the presence of conservative motifs M1–M3 (Harpke and Peterson 2008; Jobes and Thien 1997). Next, we investigated whether the secondary structures of ITS2 in Q. phakopsoricola displayed the conserved features expected for ITS2 sequences in eukaryotic organisms (Schultz et al. 2005; Coleman 2007). Prediction of the ITS2-proximal stem secondary structure was calculated on a DINAMelt web server (Markham and Zucker 2005). Secondary structures were drawn with VARNA (Darty et al. 2009).
We used the GlobalFungi database v0.9.8; Release version 2.0 (Větrovský et al. 2020; accessed 18.2. 2021) to see if ITS sequences of Q. phakopsoricola were captured by metabarcoding studies. We used ITS1 and ITS2 sequences separately and searched using the “BLAST—group results” option. Our sequences were compared to results obtained from 26,978 environmental samples from 255 published studies covering 145,873,740 unique sequence variants representing approximately 0.8 × 109 sequence reads.
Results
Morphology
The morphology of the fungicolous fungus as seen macroscopically and by light microscopy (Figs. 2, 3) was similar to that of species of Ramularia, Pezizomycotina, Ascomycota (Braun 1998), particularly of the rust-inhabiting (uredinicolous) species (Braun 1998). SEM and TEM showed surface ornaments of conidia and conidiophores (Figs. 4, 5, 7, 8) similar to those of Ramularia species as well. Conidiogenous loci in the present Ramularia-like hyphomycete, however, were flat or slightly convex in contrast to those known for Ramularia having a conical apically pointed center separated from the marginal rim by an abrupt hollow (Fig. 6). A minute pore was visible in some conidiogenous loci of the Ramularia-like hyphomycete (Fig. 5c). TEM revealed important ultrastructural details (Figs. 7, 8). Dolipore-like extensions of the septal cell wall around the pore could only be seen once in in situ fixed material (Fig. 7a), but no pores were found in the material from culture. Woronin bodies, parenthesomes, or other systematically significant details were not detected. The round electron-dense plug at the conidial base in Fig. 8a at higher magnification revealed to be composed of granular material and was not homogeneous like a Woronin body. The cell wall ornament seen by light microscopy and SEM revealed to be composed of electron-dense material superficially attached to the surface of the cell wall without bases in the underlying cell wall (Fig. 7c, 8b, c). Conidium dehiscence appeared to be schizolytic, since no empty separating cell indicating rhexolytic dehiscence was found. Conidiogenous loci and conidial hila were thickened between the ridges of the ruptured outer cell wall layer by electron-transparent material, into which a more electron-dense presumably ring-shaped structure was embedded (Figs. 7b, 8). By deposition of further electron-transparent material, the electron-dense ring appeared to be pressed downwards into the conidiogenous cell. The resulting structure resembling a one-sided dolipore (Fig. 8b, e) was not found in the conidia. The cell walls showed diffuse to distinct layers, but were less electron-transparent than the material in the conidiogenous loci and conidial hila.

Quasiramularia phakopsoricola. a White spots formed on orange rust sori (Phakopsoraceae) on the abaxial side of a leaf of Ampelopsis brevipedunculata (R. Kirschner 4400). b, c Whitish pustules formed by conidiophores and conidia (arrows) on the orange rust sori on Parthenocissus tricuspidata in situ (R. Kirschner 5313). d Petri dish (9-cm diam.) with culture (WIC009) seen from above (CMA, ca. 1 month). e Same culture as in d, reverse. Scale bars: b = 2 mm, c = 1 mm

Quasiramularia phakopsoricola, light microscopy. a Conidiophores and conidia from the host in situ. b Conidiophores and conidia formed by the fungus in culture (WIC009). Scale bars = 10 µm

Quasiramularia phakopsoricola (WIC009) on a rust fungus (Phakopsoraceae) on Ampelopsis brevipedunculata as seen by scanning electron microscopy (SEM). a Two rust sori, the right one heavily overgrown by Q. phakopsoricola. b Conidiophores among urediniospores and paraphyses. c Conidia of Q. phakopsoricola germinating on the surface of a urediniospore. Scale bars: a = 50 µm, b = 30 µm, c = 10 µm

SEM photographs showing conidiogenous cells and conidia with surface ornament, conidiogenous loci, and hila of conidia of Q. phakopsoricola (WIC009). a Conidiogenous cells with terminal geniculations and conidiogenous loci (one locus indicated by an arrow). b Fusiform conidium with hilum (arrow). c Conidiogenous cell with conidiogenous locus and a central pore (arrow). d Conidiogenous loci and one hilum of a conidium (arrows). Scale bars: a, b, d = 5 µm, c = 3 µm

SEM photographs showing three conidiogenous loci of Ramularia pusilla (R. Kirschner 3143) with the raised center clearly separated from the round rim by a ring-shaped hollow. Scale bar = 3 μm

TEM photographs of cell wall structures of Q. phakopsoricola. a Septal pore (arrow) in a hypha from material in nature (WIC009). b Conidium with surface ornament. The arrow indicates the hilum as shown in Fig. 5d (R. Kirschner 4689). c Surface ornament at higher magnification. Scale bars a = 2 μm, b = 2 μm, c = 0.2 μm

TEM photographs of conidiogenous loci of Q. phakopsoricola (R. Kirschner 4689). a Detachment of conidium base from a conidiogenous locus. Cell walls of both structures have thickened electron-transparent material (white arrowheads) between the thin electron-dense outer layer of cell wall (white arrows) which is connected to the cell wall of the entire conidiogenous cell. Note the more electron-dense (slightly shaded) presumably ring-shaped structure (black arrows) embedded in the electron-transparent material (arrows). b Conidiogenous locus with shaded area (arrowhead) embedded in electron-transparent material (white arrowhead) and extending into a presumably ring-shaped electron-dense structure (black arrows) which is projecting into the cytoplasm. c, d Strongly electron-dense presumably ring-shaped structures (black arrows) embedded in electron-transparent material (white arrowhead in d) within conidiogenous locus. e Presumably ring-shaped electron-dense structure (black arrows) appearing to be pressed by copious electron-transparent material (white arrowhead) into the cytoplasm. An electron-dense outer cell wall layer (white arrow) is continuous with the outer cell wall layer of the conidiogenous cell. Scale bars: a, c, d = 0.5 μm; b, e = 0.2 μm
Molecular data and phylogenetic analyses
BLAST searches with LSU rDNA, SSU rDNA, and RPB2 DNA sequences indicated a close relationship of the Ramularia-like hyphomycete with species belonging to Ustilaginomycotina, Basidiomycota. The closest matches of SSU rDNA sequence data presented similarity values of 90–89.5% with various species of Ustilaginomycotina (e.g., Robbauera albescens (Gokhale) Boekhout et al. NG_061194, Quambalaria cyanescens (de Hoog & G.A. de Vries) Z.W. de Beer KF706440, and Entyloma ficariae A.A. Fisch. Waldh. KP322949). Matches below 89.5% similarity belonged to species of Agaricomycetes (e.g., Rhizoctonia solani J.G. Kühn DQ917659). The same pattern was obtained for LSU rDNA sequence data for which species of Ustilaginomycotina exhibited similarity values of 90.0–91.1% (e.g., Tilletiopsis washingtonensis Nyland MH868275, Entyloma ficariae HM046476) and species of Agaricomycetes values below 90.0% (e.g., Rigidoporus microporus (Sw.) Overeem AY333795).
The ITS sequences of three strains of the Ramularia-like hyphomycete were 100% identical (R. Kirschner 4365, R. Kirschner 4689, and WIC009), but the ITS sequence of a further strain (R. Kirschner 4704) differed for 30 bp (4.2%) positions, from which two were in the highly conserved 5.8S region. The ITS alignment of four strains revealed 22 substitutions, from which seven represented RIP-like-type (C to T) changes. In the LSU sequences, the strain R. Kirschner 4707 differed in 9 bp (1.5%) positions from others, whereas SSU sequences were identical. Because the source strains have identical phenotype and ecology, we are interpreting these differences as intraspecies variability (see Discussion for further details). A similarity search using discontinuous MegaBlast in NCBI GenBank did not result in any hit in the case of ITS1 and ITS2 sequences that were analyzed separately. Search using a 5.8S rDNA sequence showed low similarity of about 92% to numerous species of Agaricomycotina (e.g., Gymnopus spongiosus (Berk. & M.A. Curtis) Halling MK575231), whereas the closest matches from Ustilaginomycotina were 87.6% (e.g., Entyloma ficariae MT644871). The ITS1 and ITS2 sequence similarity search against GlobalFungi database did not result in any reliable hit, either.
Phylogenetic analyses based on sequence data of the single gene and concatenated datasets (LSU + SSU, RPB2, LSU + SSU + RPB2) suggested that the Ramularia-like hyphomycete represents a lineage new to science within Ustilaginomycotina (Fig. 1, ESM4). Our sequences formed a long branch close to Capitulocladosporium (all analyses), Ceraceosorales (rDNA), or Malasseziales, and showed a basal position within the Ustilaginomycotina. The position of the two latter orders was incongruent between datasets and analyses with different settings (Fig. 1, ESM4). The Ramularia-like fungus is distinct with respect to morphology, DNA barcode genes, and ecology, not ascribable to already described taxa. Therefore, a new order and a new family based on the new genus and species Quasiramularia phakopsoricola are proposed below.
GC content of rDNA sequences in Basidiomycota.
Our analysis of GC contents in rDNA sequence data from numerous species of Basidiomycota showed considerable heterogeneity in GC content between and within the main lineages. Quasiramularia phakopsoricola has one of the lowest GC content values among all rDNA sequences considered in this study. Its GC values are lower than those in any of the LSU, ITS2, and SSU rDNA datasets of species of Ustilaginomycotina species and are among the lowest in ITS1 and 5.8S rDNA datasets. Its GC content is also much lower than the content values of most of the published LSU, SSU rDNA sequences of species of Agaricomycotina and Pucciniomycotina. The values of Q. phakopsoricola are similar, however, to ITS1, 5.8S, and ITS2 rDNA data of species of Pucciniomycotina and to all rDNA regions of Wallemiomycotina and Entorrhizomycetes (Fig. 9, Table S1).

Box plots with outliers summarizing the rDNA GC content of Quasiramularia and Basidiomycota sequences deposited in GenBank
GC content values strongly diverging from values of closely related species were discovered for several relationships of species in Basidiomycota. Such outlier sequences were discovered in the LSU rDNA dataset of Agaricomycotina for Entoloma virescens (Sacc.) E. Horak ex Courtec. (GU384622). For the first part of SSU rDNA, an unpublished sequence of Meira nicotianae H.K. Wang & F.C. Lin (MT860276) in the Ustilaginomycotina is a further example. For the ITS rDNA region, the outliers belonged to species of Meira (ITS1 dataset, Ustilaginomycotina), the invalidly published Reniforma strues Pore & Sorenson (ITS1 dataset, KX011609, KX015891, Pucciniomycotina), and Entoloma mengsongense Ediriweera et al. (ITS1, ITS2, KU131556, Agaricomycotina). Interestingly, among all fungi, the species of rust fungi belonging to the genus Phakopsora had low GC content values in all rDNAs, similar to those of the uredinicolous Q. phakopsoricola (Table S1). Concerning the high GC values, for all coding rDNA regions, the outliers often belong to Ceraceosorales, Gyroporus, Kurtia argillacea, Moniliellales, Malasseziales, Subulicystidium, Tulasnella, Violaceomyces, and Wolfiporia.
Secondary structure of Quasiramularia phakopsoricola rRNA.
For the ITS2 rRNA haplotypes represented by R. Kirschner 4365 and R. Kirschner 4704, the homology modeling provided by the ITS2 database did not provide any model of known ITS2 secondary structure. The direct ITS2 secondary structure prediction resulted in a structure which consisted of four helices (I–IV) radiating from a central loop and a minimum free energy of − 54.60 kcal/mol. It has an unpaired uracil at the helix II and the helix III contains the conserved motif 5′-UGUGA-3′, close to its apex (Fig. 10). Direct folding of the second ITS2 haplotype (R. Kirschner 4704) resulted in the same secondary structure, with the same conservative motives, but with lower minimum free energy (− 67.30 kcal/mol) (ESM3: Fig. S1).

Predicted secondary structures of RNAs of Quasiramularia phakopsoricola and Ustilago hordei. a 5.8S rRNA. b ITS2. c ITS2-proximal stem. The asterisks show the variable positions in the 5.8S region caused by the sequence variation in strain R. Kirschner 4704. The helixes of 5.8S and ITS2 are numbered by Roman numerals I–IV. The conservative motifs in 5.8S (M1–M3) and ITS2 are highlighted in gray
In the 5.8S rRNA, the sequence motifs universally present in angiosperms (Harpke and Peterson 2008; Jobes and Thien 1997) as M1 (5′-CGAUGAAGAACGCAGC- 3′), M2 (5′-AAUUGCAACGUUA-3′), and M3 (5′-UUUGAACGCA-3′) were detected, with differences only present in the M2 region (5′-AAUUGCAACGUUA-3′). Variability in the M2 region was also found in the M2 region of Ustilago hordei (Pers.) Lagerh. (5′- AAUUGCAGAAGUG -3′). The 5.8S rRNA sequence has two polymorphic sites within the aligned 5.8S rRNA sequences of Q. phakopsoricola, not affecting its secondary structure or sequence of conservative motifs (Fig. 10).
The ITS2-proximal stem structure formed by the 5′ end of the 28S rRNA and the 3′end of the 5.8S rRNA of Q. phakopsoricola consists of a single stem with two bulge loops which are separated from each other by 6 bp (Fig. 10).
Taxonomy
Quasiramulariales R. Kirschner, M. Kolařík & M. Piepenbr., ord. nov.
Index Fungorum IF558774.
Based on the genus Quasiramularia I-Chin Wei & R. Kirschner, in opere ipso.
Diagnosis: Belonging to Ustilaginomycotina, anamorph morphologically similar to anamorphic Ramularia Unger, on solid substrates budding yeast cells and slimy colonies absent.
Quasiramulariaceae R. Kirschner, M. Kolařík & M. Piepenbr., fam. nov.
Index Fungorum IF558773.
Based on the genus Quasiramularia I-Chin Wei & R. Kirschner, in opere ipso.
Diagnosis: Belonging to Ustilaginomycotina, anamorph morphologically similar to anamorphic Ramularia Unger, on solid substrates budding yeast cells and slimy colonies absent.
Quasiramularia I-Chin Wei & R. Kirschner, gen. nov.
Index Fungorum IF558771.
Etymology: Composed from quasi (Latin: similar) and the genus name Ramularia.
Typus generis: Quasiramularia phakopsoricola I-Chin Wei & R. Kirschner, in opero ipso.
Diagnosis: Similar to anamorphic Ramularia Unger, but belonging to the Ustilaginomycotina and conidiogenous loci lacking the ring-shaped hollow between the central dome and the marginal ring typical of Ramularia species as seen by scanning electron microscopy.
Quasiramularia phakopsoricola I-Chin Wei & R. Kirschner, sp. nov. Figures 2–5, 7–8
Index Fungorum IF558772.
Etymology: Referring to the genus of the host fungus, Phakopsora.
Hyphae and conidiophores formed in white to dirty white dusty-dry pustules on rust uredinia on the abaxial side of living leaves. Conidiophores emerging from uredinia, loose, erect to decumbent, effuse, more or less straight, subcyclindric, septate, hyaline or subhyaline, smooth to finely verruculose, simple or with few short branches, (13–)22–44(–60) × 2–3(–4) µm (n = 30). Conidiogenous cells mostly terminal, rarely subterminal or lateral, subcyclindric or slightly swollen, (4–)10–22(–25) × 2–3 µm (n = 30); conidiogenous loci thickened and slightly darkened. Conidia catenate, ellipsoid-ovoid or subcylindric-fusiform, ends rounded to somewhat pointed, (6–)7–15(–20) × 4–5(–7) µm (n = 30), hyaline or subhyaline, smooth to rough, hila thickened, slightly darkened, less than 1-μm diam.
Colonies growing slowly on CMA at room temperature, reaching approx. 25-mm diam. after 1 month, whitish, subeffuse to powdery on top side, dark greenish pigment released on reverse side. Conidiophores similar to those on the host, (42–)49–131(–150) × 2 μm (n = 30). Conidiogenous cells subcyclindric, 5–19(–22) × 2 µm. Conidia catenate, ellipsoid-ovoid, fusiform, or subcylindric-fusiform, ends rounded to somewhat pointed, (7–)10–18(–23) × 3–5(–6) µm (n = 30), hyaline, smooth to rough, hila thickened, slightly darkened.
Specimens examined (dried specimens on natural substrate, if not stated otherwise): On Phakopsora ampelopsidis Dietel & P. Syd. (Phakopsoraceae) on Ampelopsis brevipedunculata (Vitaceae), Taiwan, Taoyuan City, Zhongli District, north of National Central University, Neicuo 3rd Rd., 24.97510107, 121.19031800, ca. 135 m, 10. Nov 2015, I.C. Wei & R. Kirschner WIC009 (TNM, holotype), ex-type strain BCRC FU30724, ITS MT741740, SSU MT741743, LSU MT741723; ibid., 04. Dec 2015, I.C. Wei WIC018 (TNM); ibid., 28. Dec 2015, I.C. Wei WIC019A (on leaves) and WIC019B (dried culture) (TNM); ibid., 20. Mar 2017, R. Kirschner 4383 (TNM); New Taipei City, Yingge District, Yingge Rock hiking trail, ca. 24.958814, 121.360379, ca. 80 m, 23. May 2017, R. Kirschner 4400 (TNM); ibid., 27. Oct 2018, R. Kirschner 4689 (TNM); Taipei City, Zhongshan District, Jian Butterfly Garden, 25.086900, 121.554600, ca. 350 m, 01. Feb 2017, R. Kirschner 4365 (TNM), strain BCRC FU30777, ITS MT741738, SSU MT741741, LSU MT741721, RPB2 LC535131. On Ph. ampelopsidis on Parthenocissus tricuspidata (Siebold & Zucc.) Planch. (Vitaceae), Taipei City, Daan District, border of Fuzhoushan Park at Wolong Street, ca. 25.017834, 121.552391, ca. 10 m, 31. Mar 2019, R. Kirschner 4704 (TNM), ITS MT741739, SSU MT741742, LSU MT741722, RPB2 LC535132, ibid., 19. Aug 2021, R. Kirschner 5313 (TNM).
Discussion
Morphology and taxonomic identity
Morphologically, the fungus growing on a rust in Taiwan presented here is indistinguishable from Ramularia, which contains species growing on rust fungi, namely R. coleosporii Sacc., R. uredinicola Khodap. & U. Braun, R. uredinearum Hulea, and R. uredinis (W. Voss) Sacc. (Braun 1998; Khodaparast and Braun 2005; Bakhshi et al. 2021). Hitherto, however, no member of the rust family Phakopsoraceae has been recorded as host of fungicolous Ramularia species. Since sizes of conidiophores and conidia also differ slightly from the known Ramularia species on rust fungi and conidia consistently lack septa in contrast to those of the known uredinicolous species, we can exclude that the taxon from Taiwan was described as a species of Ramularia in the past.
Some anamorphs of Ustilaginomycotina are morphologically similar to Ramularia species and were described as species of Ramularia or its synonym Ovularia in the past, but they lack the darkened or thickened conidiogenous loci and hila of the species presented here (Braun 1995, 1998). Quambalaria pitereka (J. Walker & Bertus) J.A. Simpson was first described as Ramularia species (Walker & Bertus 1971) and was transferred to Sporothrix (Braun 1998), before its affiliation to Ustilaginomycotina was assumed (Simpson 2000) and confirmed by DNA sequence analysis (de Beer et al. 2006). The basidiomycetous nature of Quambalaria species was further confirmed by TEM by the discovery of dolipores (de Beer et al. 2006). Quambalaria species are plant parasitic or associated with insects and form smooth conidia and a yeast stage (Simpson 2000; Kolařík et al. 2006; Antropova et al. 2014). A teleomorph is not yet known. Another recently proposed genus without known teleomorph, Capitulocladosporium, was proposed for a hyaline or faintly pale brown Cladosporium-like species isolated from a midge (Sun et al. 2018). By its undarkened conidiogenous loci and conidial hila, it differs from Cladosporium. Like in Quasiramularia, a yeast stage is absent also in Capitulocladosporium (Sun et al. 2018). The conidiophores of Capitulocladosporium form many coronate conidiogenous loci on the strongly swollen apex of the conidiophore, whereas the not or only slightly swollen conidiophore apex in Quasiramularia bears only a few conidiogenous loci. Conidiophores and conidia are smooth in Capitulocladosporium (Sun et al. 2018) but carry distinct ornaments in Quasiramularia. Some yeast-like anamorphs belonging to Anthracocystis, Moesziomyces, Pseudozyma, Robbauera, Tilletiopsis, and Ustilago (including Mycosarcoma) have been considered as colonizers of powdery mildews (Kiss 2003). Among uredinicolous fungi, Quasiramularia seems to be the first representative from Ustilaginomycotina. These examples together with the fungicolous Q. phakopsoricola indicate a shift of several anamorphs of Ustilaginomycotina to chitinous substrates (other fungi, arthropods). Further plant-parasitic anamorphic Ustilaginomycotina and Ramularia or Ovularia confused in the past (Braun 1995) are now to be treated under their respective teleomorph names, such as Entylomella under Entyloma (Aime et al. 2018). Although Quasiramularia represents an ecologically and phylogenetically new lineage, we hesitated to provide new names above the genus rank. A single species and the absence of phylogenetically distinct markers other than mere DNA sequence deviations do not seem to warrant new names above the genus level. Since inflation of taxonomy with such names apparently has become an inevitable practice, however, we decided to propose the new family and order here.
The ultrastructure of septal pores in Ustilaginomycotina is diverse. Membranous pore caps of simple pores occur in most Ustilaginomycotina but septal pores can also be missing as in the Georgefischeriales and Ustilaginales (Bauer et al. 1997; Oberwinkler and Bauer 2018). Species of Urocystidales are characterized by non-membranous bands in the pores. Pores are mostly simple, except for Tilletiales and Quambalariaceae within the Microstromatales that have dolipores (Bauer et al. 1997; de Beer et al. 2006; Oberwinkler and Bauer 2018). We could find a dolipore-like structure in a vegetative hyphal septum only once in the material from nature. In the conidiogenous loci, a structure looking like a half-sided dolipore was occasionally seen. Its central area was distinctly separated from the cytoplasm and did not form a channel that could connect the cytoplasm of the conidiogenous cell and the conidium. Furthermore, there was no corresponding mirroring structure in the conidium base.
The cell wall structure and ornamentation were rather unspecific, since similar electron dense cones superficially attached to the outer cell wall were also found in anamorphic ascomycetous and pucciniomycetous fungi (Hammill 1973; Kirschner et al. 2002). The ornamentation is lacking in the TEM photographs of the ustilaginomycetous Quambalaria (Antropova et al. 2014). The innermost cell wall layer(s), however, were always more electron-dense than the electron-transparent material in the conidiogenous loci and conidial hila. This cell wall ultrastructure corresponds to that known for many Basidiomycota, whereas in Ascomycota, the innermost layer usually is the most electron-transparent part of the cell wall (Kirschner and Oberwinkler 2001). Conidiogenesis has rarely been investigated in anamorphic Ustilaginomycotina. The ultrastructure of sympodial conidiogenesis among Ustilaginomycotina was studied in detail for Exobasidium vaccinii (Fuckel) Woronin by Mims and Richardson (1987). Although not mentioned in the text, copious electron-transparent material in the conidiogenous locus as well as in the corresponding conidial hilum was clearly illustrated (Mims and Richardson 1987). A central thin electron-dense isthmus in the conidiogenous locus is visible in some TEM photographs, but not an electron-dense ring-like structure within the electron-transparent material as in Q. phakopsoricola. The conidiogenous loci and hila in Quambalaria appear homogenously electron-dense in the TEM photographs of Antropova et al. (2014). Copious electron-transparent material is also characteristic for Tritirachium roseum J.F.H. Beyma (Pucciniomycotina), but a ring-shaped structure inside this material could not be detected (Hammill 1973). The accumulation of electron-transparent material prior to conidium dehiscence may be a widespread feature of anamorphic Basidiomycota with sympodial conidium development, but ultrastructural characteristics of many further species belonging to different groups of Ascomycota and Basidiomycota are necessary to evaluate the phylogenetic significance.
We did not find TEM illustrations of conidiogenous loci of other species of Ustilaginomycotina or Ramularia in literature. Cercospora and Ramularia are fairly closely related and both have strongly darkened conidiogenous loci visible by light microscopy. For conidiogenous loci and conidial hila in Cercospora, rather unspecific granular electron-dense material between the ridges of the ruptured outer cell wall layer appears predominant (Pons et al. 1985). To our knowledge, the presence of an electron-dense ring within copious electron-transparent material in the conidiogenous loci and conidial hila as in Q. phakopsoricola has not been reported in other fungi.
rDNA features in Quasiramularia phakopsoricola and other Basidiomycota.
The ITS sequences of Quasiramularia phakopsoricola have very low GC content values, and similar values were rarely found among so-far published sequences. A chimeric nature can be excluded, because the identical sequences were obtained from independent DNA extracts and several specimens. The low GC content and very unique sequence are typical signs of a pseudogene (see Kolařík and Vohník 2018 and Stadler et al. 2020 for review).
rDNA pseudogenes have irregular 5.8S rDNA sequences and lack some or all of the conserved regions typically present in ITS2 rDNA sequences (Coleman 2007; Hunter et al. 2007; Kocot and Santos 2009; Glass et al. 2013).
ITS2 rRNA secondary structures typically present four helices (Fig. 10). Helix II is usually composed of fewer than 12 base pairs, is never branched, and contains a U–U bulge at its base (Schultz et al. 2005; Coleman 2007). Helix III typically is the longest helix and contains a distinctive motif close to its apex (Schultz et al. 2005). In the ITS secondary structure of Q. phakopsoricola, four helices were detected and helix III contains the motif 5′-UGUG-3′, which may have replaced the conserved motif 5′-UGGU-3′ which is typical for fungi (Schultz et al. 2005; Freire et al. 2012). Q. phakopsoricola also presents an unpaired uracil in helix II, instead of a U-U mismatch, which is known for some fungi (Glass et al. 2013). The observed pattern of the secondary structure of the ITS molecule of Q. phakopsoricola is similar to the pattern of secondary structures of functional ITS2 known in other fungi (Freire et al. 2012; Glass et al. 2013; Rampersad 2014; Kapoor et al. 2018).
In the 5.8S gene (M1, M2, M3), at least three motifs are conserved among angiosperms (Harpke and Peterson 2008) and the same or slightly modified sequence strings occur in fungi. In total, across all three regions, a single substitution change is reported in Colletotrichum (Rampersad 2014), two in Induratia (“Muscodor”, Kapoor et al. 2018), three in Batrachochytrium dendrobatidis Longcore, Pessier & D.K. Nichols (Glass et al. 2013), and four in Phakopsora (Freire et al. 2012) and Conidiobolus (Glass et al. 2013). This variability is similar to the variability observed for Q. phakopsoricola.
Finally, we studied the hybridization model of the ITS2-proximal stem, the so-called 5.8S–28S rRNA proximal stem. The structure of this stem is very conservative, and any deviation from this structure is a reliable sign of the presence of pseudogenes (Harpke and Peterson 2008). The ITS2-proximal stem typically has two unpaired nucleotides, one on each side, with approximately six base pairs in between, resulting in the formation of two bulge loops. In our case, the ITS2-proximal stem structure corresponds to the structure typical for eukaryotes and Saccharomyces cerevisiae (Desm.) Meyen (Peculis and Greer 1998) (Fig. 10). Given these important arguments, we conclude that the ITS sequences of Q. phakopsoricola are functional gene copies. rDNA sequences with low GC content can be functional, as was shown for rust fungi (Phakopsora; Freire et al. 2012).
Our study is the first survey of GC content values across a large set of rDNA sequence data of Basidiomycota. We discovered that low rDNA GC content is typical for many species of Pucciniomycotina and all species of Wallemiomycetes and Entorrhizomycetes. The GC content of DNA sequences is shaped by a complex balance between mutation, selection, recombination, and genetic drift. As a consequence of variation in this subtle balance, it has been observed that GC content varies considerably among different species and along chromosomes of a single species (reviewed by Foerstner et al. 2005; Romiguier and Roux 2017) often forming distinct GC-rich and GC-poor regions (Testa et al. 2016). Several hypotheses have been proposed to explain the origin and evolution of GC content variation. Typical intrinsic molecular processes include transcriptional machinery with mutational bias (McLean and Tirosh 2011), GC-biased gene conversion (gBGC) (Pessia et al. 2012), and repeat-induced point mutation (RIP). RIP is only present in sexual organisms, known to be responsible for the origin of GC-poor regions, and considered to act primarily as a defense against transposon propagation. RIP targets repetitive DNA and decreases GC content through the conversion of cytosine to thymine bases. RIP has been identified as a driver of fungal genome evolution, as RIP can also occur in single-copy genes neighboring repeat-rich regions (Costantini et al. 2013; Testa et al. 2016) and rDNA tandem repeats (Rouxel et al. 2011). Consequently, in the so-called “two speed” genome concept, the coding genes located in repeat rich, GC-poor regions tend to have higher evolutionary rates than genes in GC-rich regions (Wang et al. 2017). However, in Quasiramularia, we did not recognize typical RIP-induced mutations and the presence of RIP would not conform to the tentative asexuality of this fungus. RIP, gBGC, and other genetic particularities are somehow linked to life history changes affecting population size and the frequency of sexual recombination. Population subdivision, recurrent bottlenecks, and effective asexuality related to lifestyle changes (e.g., the transition from free-living lifestyles to obligate host-associated forms) are probably responsible for decrease in GC content in bacteria (McCutcheon and Moran 2012) and Microsporidia (Haag et al. 2020). This can be accompanied by increased mutation rates as was shown in endosymbiotic (Woolfit and Bromham 2003) and pathogenic fungi (Bromham et al. 2013; Fillinger and Anderson 2019). Finally, the transition from saprobes to symbionts and pathogens can be accompanied by a shift towards higher proportions of GC-poor regions, which have higher mutation rates (Testa et al. 2016). According to the phylogenetic hypotheses presented in this study, Q. phakopsoricola probably evolved from saprotrophic, facultative plant parasitic fungi and transformed into a colonizer of a specific rust species. Q. phakopsoricola lives in strongly fragmented niches, which can accelerate the mutation rate via effects of small effective population size and repeated bottlenecks (Woolfit and Bromham 2003, 2005; Lynch 2010). A switch to asexuality, if present in Q. phakopsoricola, can have the same effect on the mutation rate as was seen in many organisms (Bast et al. 2018). This might explain GC content changes and fast mutation rates in Q. phakopsoricola, but also the very high intraspecific variability in rDNA sequences.
Compositional biases in DNA sequences adversely affect phylogenetic analyses (reviewed in Romiguier and Roux 2017). This can be partially solved by the use of sequence evolution models that take into account heterogeneity in GC content and substitution rates (i.e., heterotachy). The success of such approaches, however, is limited in the case of highly GC-heterogeneous datasets and in sequences with high degrees of sequence saturation (Betancur et al. 2013; Romiguier et al. 2016). Protein sequences may yield more reliable results because they are less affected by nucleotide biases than rDNA sequences (Foster and Hickey 1999).
Conclusions
The new species Quasiramularia phakopsoricola is a specific colonizer of rust fungi, which is a unique lifestyle among Ustilaginomycotina. We assume that this fungus performed a dramatic lifestyle switch during its evolution, which might explain changes in the GC content. GC content values of Q. phakopsoricola are similar to those of its rust host, but far from its taxonomic relatives. The same processes resulting in higher substitution rates might explain the presence of unique ITS sequences. For phylogenetic analyses, we, therefore, do not use ITS sequence data of Q. phakopsoricola, which would cause artificial results of the analyses. We kept the analyses of rDNA and protein gene sequence data separate, because a mixing of relatively long, GC-biased rDNA markers with more informative, but shorter protein coding genes results in phylogenetic artifacts. This should be taken into account for taxa which are placed on long branches in the phylogenetic trees and have high (Kolařík and Vohník 2018, see Table S1 for other taxa) or low GC content (Table S1), but which are closely related according to other genetic markers or according to morphological and ecological characteristics. This is at least partially valid for Entorrhizomycetes and Golubevia pallescens (Gokhale) Q.M. Wang et al. (nom. inval., Ustilaginomycetes), Ceraceosorales (Wang et al. 2015a), and Reniforma strues (nom. inval., Pucciniomycotina, Aime et al. 2006, Wang et al. 2015b). Long branch formation evidently due to GC bias can be demonstrated for the outliers in Agaricomycotina (Fig. 9), e.g., Entoloma mengsongense (Ediriweera et al. 2017), Limacella bangladeshana (nom. inval., KR816866, Hosen and Li 2017), Subulicystidium oberwinkleri (Ordynets et al. 2018), Tulasnella calospora (Cruz et al. 2011), Wolfiporia (AF082671, Km and Jung 2001), and for the Malasseziales and Entorrhizales (Begerow et al. 2006).
Data availability
All data and materials are provided.
Code availability
Not applicable.
References
Aime MC, Matheny PB, Henk DA, Frieders EM, Nilsson RH, Piepenbring M, McLaughlin DJ, Szabo LJ, Begerow D, Sampaio JP (2006) An overview of the higher level classification of Pucciniomycotina based on combined analyses of nuclear large and small subunit rDNA sequences. Mycologia 98:896–905
Aime MC, Castlebury LA, Abbasi M, Begerow D, Berndt R, Kirschner R, Marvanová L, Ono Y, Padamsee M, Scholler M, Thines M, Rossman AY (2018) Competing sexual and asexual generic names in Pucciniomycotina and Ustilaginomycotina (Basidiomycota) and recommendations for use. IMA Fungus 9:75–89
Antropova AB, Bilanenko EN, Mokeeva VL, Chekunova LN, Kachalkin AV, Shtaer OV, Kamzolkina OV (2014) Report of Quambalaria cyanescens in association with the Birch (Betula pendula). Microbiology 83(5):690–698
Bakhshi M, Zare R, Jafary H (2021) Identification of Acrodontium luzulae and Ramularia coleosporii, two fungicolous fungi on Melampsora hypericorum, the causal agent of Hypericum androsaemum rust. Nova Hedwigia 113(3–4):323–338. https://doi.org/10.1127/nova_hedwigia/2021/0652.
Bast J, Parker DJ, Dumas Z, Jalvingh KM, Van Tran P, Jaron KS, Figuet E, Brandt A, Galtier N, Schwander T (2018) Consequences of asexuality in natural populations: insights from stick insects. Mol Biol Evol 35:1668–1677
Bauer R, Oberwinkler F, Vánky K (1997) Ultrastructural markers and systematics in smut fungi and allied taxa. Can J Bot 75:1273–1314
Begerow D, Stoll M, Bauer R (2006) A phylogenetic hypothesis of Ustilaginomycotina based on multiple gene analyses and morphological data. Mycologia 98:906–916
Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sánchez-García M, Ebersberger I, de Sousa F (2013) Improved software detection and extraction of ITS1 and ITS 2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 4:914–919
Betancur-R R, Li C, Munroe TA, Ballesteros JA, Ortí G (2013) Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes). Syst Biol 62:763–785
Braun U (1995) A monograph of Ramularia, Cercosporella and allied genera (phytopathogenic hyphomycetes), vol I. IHW-Verlag, Eching, Germany
Braun U (1998) A monograph of Ramularia, Cercosporella and allied genera (phytopathogenic hyphomycetes), vol II. IHW-Verlag, Eching, Germany
Bromham L, Cowman PF, Lanfear R (2013) Parasitic plants have increased rates of molecular evolution across all three genomes. BMC Evol Biol 13:1–11
Coleman AW (2007) Pan-eukaryote ITS2 homologies revealed by RNA secondary structure. Nucl Acids Res 10:3322–3329
Collins TM, Fedrigo O, Naylor GJP (2005) Choosing the best genes for the job: the case for stationary genes in genome-scale phylogenetics. Syst Biol 54:493–500
Costantini M, Alvarez-Valin F, Costantini S, Cammarano R, Bernardi G (2013) Compositional patterns in the genomes of unicellular eukaryotes. BMC Genomics 14:1–12
Cruz D, Suárez JP, Kottke I, Piepenbring M, Oberwinkler F (2011) Defining species in Tulasnella by correlating morphology and nrDNA ITS-5.8S sequence data of basidiomata from a tropical Andean forest. Mycol Prog 10:229–238
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772
Darty K, Denise A, Ponty Y (2009) Varna: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25:1974–1975
De Beer ZW, Begerow D, Bauer R, Pegg GS, Crous PW, Wingfield MJ (2006) Phylogeny of the Quambalariaceae fam. nov., including important Eucalyptus pathogens in South Africa and Australia. Stud Mycol 55:289–298
Ediriweera AN, Karunarathna SC, Xu J, Hyde KD, Mortimer PE (2017) Entoloma mengsongense sp. nov. (Entolomataceae, Agaricales), a remarkable blue mushroom from Yunnan Province. China Turkish Journal of Botany 41:505–515
Fillinger RJ, Anderson MZ (2019) Seasons of change: mechanisms of genome evolution in human fungal pathogens. Infect Genet Evol 70:165–174
Foerstner KU, von Mering C, Hooper SD, Bork P (2005) Environments shape the nucleotide composition of genomes. EMBO Rep 6:1208–1213
Foster PG, Hickey DA (1999) Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J Mol Evol 48:284–290
Freire MCM, da Silva MR, Zhang X, Almeida ÁMR, Stacey G, de Oliveira LO (2012) Nucleotide polymorphism in the 5.8 S nrDNA gene and internal transcribed spacers in Phakopsora pachyrhizi viewed from structural models. Fungal Genet Biol 49:95–100
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes–application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118
Glass DJ, Takebayashi N, Olson LE, Taylor DL (2013) Evaluation of the authenticity of a highly novel environmental sequence from boreal forest soil using ribosomal RNA secondary structure modeling. Mol Phylogenet Evol 67:234–245
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Haag KL, Pombert J-F, Sun Y, de Albuquerque NRM, Batliner B, Fields P, Lopes TF, Ebert D (2020) Microsporidia with vertical transmission were likely shaped by nonadaptive processes. Genome Biol Evol 12:3599–3614
Hammill TC (1973) Fine structure of conidiogenesis in the holoblastic, sympodial Tritirachium roseum. Can J Bot 51:2033–2036
Hane JK, Oliver RP (2008) RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinformatics 9:1–12
Harpke MP, Peterson A (2008) 5.8S motifs for the identification of pseudogenetic ITS regions. Botany 86:300–305
Hofacker IL, Fontana W, Stadler PF, Bonhoeffer SL, Tacker M, Schuster P (1994) Fast folding and comparison of RNA secondary structures. Monatsh Chem 125:167–188
Hosen MI, Li T-H (2017) First report of Limacella from Bangladesh, with a new species description. Phytotaxa 332:280–286
Hunter RL, LaJeunesse TC, Santos SR (2007) Structure and evolution of the rDNA internal transcribed spacer (ITS) region 2 in the symbiotic dinoflagellates (Symbiodinium, Dinophyta). J Phycol 43:120–128
Jobes DV, Thien LB (1997) A conserved motif in the 5.8S ribosomal RNA (rRNA) gene is a useful diagnostic marker for plant internal transcribed spacer (ITS) sequence. Plant Mol Biol Rep 15:326–334
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Kocot KM, Santos SR (2009) Secondary structural modeling of the second internal transcribed spacer (ITS2) from Pfiesteria-like dinoflagellates (Dinophyceae). Harmful Algae 8:441–446
Kapoor N, Gambhir L, Saxena S (2018) Secondary structure prediction of ITS rRNA region and molecular phylogeny: an integrated approach for the precise speciation of Muscodor species. Annals Microbiol 68:763–772
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Khodaparast A, Braun U (2005) Ramularia uredinicola – a new species from Iran. Mycotaxon 91:357–359
Kirschner R (2009) Cercosporella and Ramularia. Mycologia 101:110–119
Kirschner R, Oberwinkler F (2001) Mycoparasitism by three species of Diplococcium (Hyphomycetes). Plant Biol 13:449–454
Kirschner R, Braun U, Chen Z-C, Oberwinkler F (2002) Pleurovularia, a new genus of hyphomycetes proposed for a parasite on leaves of Microstegium sp. (Poaceae). Mycoscience 43:15–20
Kiss L (2003) A review of fungal antagonists of powdery mildews and their potential as biocontrol agents. Pest Manag Sci 59:475–483
Km S-Y, Jung HS (2001) Phylogenetic relationships of the Polyporaceae based on gene sequences of nuclear small subunit ribosomal RNAs. Mycobiol 29:73–79
Kolařík M, Vohník M (2018) When the ribosomal DNA does not tell the truth: the case of the taxonomic position of Kurtia argillacea, an ericoid mycorrhizal fungus residing among Hymenochaetales. Fungal Biol 122:1–18
Kolařík M, Sláviková E, Pažoutová S (2006) The taxonomic and ecological characterisation of the clinically important heterobasiodiomycete Fugomyces cyanescens and its association with bark beetles. Czech Mycol 58(1–2):81–98
Kurtzman CP, Robnett CJ (1997) Identification of clinically important ascomycetous yeasts based on nucleotide divergence in the 5′end of the large-subunit (26S) ribosomal DNA gene. J Clin Microbiol 35(5):1216–1223
Liu YJ, Whelen S, Hall BD (1999) Phylogenetic relationships among ascomycetes: evidence from an RNA polymerase II subunit. Mol Biol Evol 16:1799–1808
Lynch M (2010) Evolution of the mutation rate. Trends Genet 26:345–352
Markham NR, Zuker M (2005) DINAMelt web server for nucleic acid melting prediction. Nucl Acids Res 33:W577–W581. https://doi.org/10.1093/nar/gki591
McCutcheon JP, Moran NA (2012) Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10:13–26
McLean MA, Tirosh I (2011) Opposite GC skews at the 5′and 3′ends of genes in unicellular fungi. BMC Genomics 12:1–10
McTaggart A, Prychid C, Bruhl J, Shivas R (2020) The PhyloCode applied to Cintractiellales, a new order of smut fungi with unresolved phylogenetic relationships in the Ustilaginomycotina. Fungal Systematics and Evolution 6:55
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Mims CW, Richardson EA (1987) An ultrastructural study of the asexual spores of the plant pathogenic fungus Exobasidium vaccinii. Bot Gaz 148(2):228–234
Oberwinker F, Bauer R (2018) Ultrastructure in basidiomycetes – requirement for function. In: P. Blanz (ed.): Biodiversity and ecology of fungi, lichens, and mosses. Kerner von Marilaun Workshop 2015 in memory of Josef Poelt. Austrian Academy of Sciences Press, Biosyst Ecol Ser 34:381–418
Ordynets A, Scherf D, Pansegrau F, Denecke J, Lysenko L, Larsson K-H, Langer E (2018) Short-spored Subulicystidium (Trechisporales, Basidiomycota): high morphological diversity and only partly clear species boundaries. MycoKeys 35:41–99
Peculis BA, Greer CL (1998) The structure of the ITS2-proximal stem is required for pre-rRNA processing in yeast. RNA 4:1610–1622
Pessia E, Popa A, Mousset S, Rezvoy C, Duret L, Marais GA (2012) Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol 4(7):675–682
Pons N, Sutton BC, Gay JL (1985) Ultrastructure of conidiogenesis in Cercospora beticola. Trans Br Mycol Soc 85(3):405–416
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Rampersad SN (2014) ITS1, 5.8 S and ITS2 secondary structure modelling for intra-specific differentiation among species of the Colletotrichum gloeosporioides sensu lato species complex. Springerplus 3:1–10
Reynolds ES (1963) The use of lead citrate at high pH as an electron opaque stain in electron microscopy. J Cell Biol 17:208–212
Riess K, Schön ME, Ziegler R, Lutz M, Shivas RG, Piątek M, Garnica S (2019) The origin and diversification of the Entorrhizales: deep evolutionary roots but recent speciation with a phylogenetic and phenotypic split between associates of the Cyperaceae and Juncaceae. Org Divers Evol 19:13–30
Romiguier J, Cameron SA, Woodard SH, Fischman BJ, Keller L, Praz CJ (2016) Phylogenomics controlling for base compositional bias reveals a single origin of eusociality in corbiculate bees. Mol Biol Evol 33:670–678
Romiguier J, Roux C (2017) Analytical biases associated with GC-content in molecular evolution. Front Genet 8:16
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rouxel T, Grandaubert J, Hane J, Hoede C, Wouw A, Couloux A, Dominguez Del Angel V, Anthouard V, Bally P, Bourras S, Cozijnsen A, Ciuffetti L, Degrave A, Dilmaghani A, Duret L, Fudal I, Goodwin S, Gout L, Glaser N, Howlett B (2011) Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun 2:202
Schultz J, Maisel S, Gerlach D, Muller T, Wolf M (2005) A common core of secondary structure on the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 11:361–364
Selig C, Wolf M, Muller T, Dandekar T, Schultz J (2008) The ITS2 Database II: homology modeling RNA structure for molecular systematic. Nucl Acids Res 36:D377–D380
Simpson JA (2000) Quambalaria, a new genus of eucalypt pathogens. Australasian Mycologist 19(2):57–62
Spurr AR (1969) A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastr Res 26:31–34
Stadler M, Lambert C, Wibberg D, Kalinowski j, Cox RJ, Kolařík M, Kuhnert E, (2020) Intragenomic polymorphisms in the ITS region of high-quality genomes of the Hypoxylaceae (Xylariales, Ascomycota). Mycol Prog 19:235–245
Sun L-Y, Sun X, Guo L-D (2018) Capitulocladosporium clinodiplosidis gen. et sp. nov., a hyphomyceteous ustilaginomycete from midge. Mycol Prog 17:307–318
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577
Testa AC, Oliver RP, Hane JK (2016) OcculterCut: a comprehensive survey of AT-rich regions in fungal genomes. Genome Biol Evol 8:2044–2064
Větrovský T, Baldrian P, Morais D (2018) SEED 2: a user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics 34:2292–2294
Větrovský T, Morais D, Kohout P, Lepinay C, Algora C, Awokunle Hollá S, Bahnmann BD, Bílohnědá K, Brabcová V, D’Alò F, Human ZR, Jomura M, Kolařík M, Kvasničková J, Lladó S, López-Mondéjar R, Martinović T, Mašínová T, Meszárošová L, Michalčíková L, Michalová T, Mundra S, Navrátilová D, Odriozola I, Piché-Choquette S, Štursová M, Švec K, Tláskal V, Urbanová M, Vlk L, Voříšková J, Žifčáková L, Baldrian P (2020) GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Scientific Data 7:228
Wei I, Kirschner R (2017) Two fungicolous anamorphic species of Hypomyces s. lat. from Taiwan. Fungal Science 32:15–25
Walker J, Bertus AL (1971) Shoot blight of Eucalyptus spp. caused by an undescribed species of Ramularia. Proceedings of the Linnean Society of New South Wales 96(2):108–115 + plates XII and XIII
Wang Q-M, Begerow D, Groenewald M, Liu X-Z, Theelen B, Bai F-Y, Boekhout T (2015) Multigene phylogeny and taxonomic revision of yeasts and related fungi in the Ustilaginomycotina. Stud Mycol 81:55–83
Wang Q-M, Groenewald M, Takashima M, Theelen B, Han P-J, Liu X-Z, Boekhout T, Bai F-Y (2015) Phylogeny of yeasts and related filamentous fungi within Pucciniomycotina determined from multigene sequence analyses. Stud Mycol 81:27–53
Wang Q, Jiang C, Wang C, Chen C, Xu J-R, Liu H (2017) Characterization of the two-speed subgenomes of Fusarium graminearum reveals the fast-speed subgenome specialized for adaption and infection. Front Plant Sci 8:140
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Woolfit M, Bromham L (2003) Increased rates of sequence evolution in endosymbiotic bacteria and fungi with small effective population sizes. Mol Biol Evol 20:1545–1555
Woolfit M, Bromham L (2005) Population size and molecular evolution on islands. Proc R Soc B 272:2277–2282
Xia X, Xie Z (2001) DAMBE: software package for data analysis in molecular biology and evolution. J Hered 92:371–373
Acknowledgements
We thank Y.-H. Yeh, NTU (Taiwan), for technical assistance with DNA methods in the laboratory and M. Basoglu, Goethe University (Germany), for generous help with TEM. We thank P. Kirk (Index Fungorum) for explaining “automatic typification” above the genus rank.
Funding
The study was supported by the Ministry of Science & Technology, Taiwan (NSC 102–2621–B–008–001–MY3 and MOST 109–2621-B-002–004).
Author information
Authors and Affiliations
Contributions
Methodology and writing concerning rDNA features such as phylogeny, GC content, and ITS features: Miroslav Kolařík. Formal analysis and investigation particularly of morphology for master thesis: I-Chin Wei. Methodology of electron microscopy: Sung-Yuan Hsieh. Writing—review and editing as well as resources in Germany: Meike Piepenbring. Conceptualization and supervision of study, funding acquisition, collection and deposit of materials, ultrastructure of conidiogenesis, writing the original draft: Roland Kirschner.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Section editor: Marc Stadler
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11557_2021_1749_MOESM2_ESM.xlsx
Supplementary file2 Table S1. GC content of ITS1, 5.8S, ITS2, LSU and SSU of Quasiramularia and other sequences from GenBank belonging to Basidiomycota. (XLSX 30837 KB)
11557_2021_1749_MOESM3_ESM.pptx
Supplementary file3 Figure S1. Secondary structure prediction of ITS2 rDNA sequence from Q. phakopsoricola R. Kirschner 4704. (PPTX 334 KB)
11557_2021_1749_MOESM4_ESM.docx
Supplementary file4 Table of DNA sequences used for the phylogenetic analyses and alternative phylogenetic trees. (DOCX 296 KB)
11557_2021_1749_MOESM5_ESM.rar
Supplementary file5 Datasets of rDNA sequences of Quasiramularia and other sequences from GenBank belonging to Basidiomycota used for the GC content calculation (RAR 5508 KB)
Rights and permissions
About this article

Cite this article
Kolařík, M., Wei, IC., Hsieh, SY. et al. Nucleotide composition bias of rDNA sequences as a source of phylogenetic artifacts in Basidiomycota—a case of a new lineage of a uredinicolous Ramularia-like anamorph with affinities to Ustilaginomycotina. Mycol Progress 20, 1553–1571 (2021). https://doi.org/10.1007/s11557-021-01749-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-021-01749-x