
Abstract
Phytophthora colocasiae is an important pathogen of taro and is widely distributed. Our goal was to develop whole genome sequence and single nucleotide polymorphism (SNP) markers to characterize historical and current populations from Hawaii (2010 and 2016, HA), historical isolates from Vietnam and China (2010, VN and CH) and current isolates from Nepal (2016, NEP). Seven isolates (VN = 2, CH = 1, HA = 1, NEP = 3) were sequenced (NCBI BioProject PRJNA378784) and compared using the reference genome of the closely related vegetable pathogen P. capsici. Genome-wide SNP analysis using 27,537 markers revealed genomes of diploid, triploid, tetraploid and higher ploidy. Ploidy varied within and between populations, with HA being primarily diploid, CH primarily triploid, VN containing diploid and triploid isolates, and NEP having predominantly triploid, tetraploid and higher ploidy. A total of 37 SNP markers were genotyped in 89 samples (grown in culture or directly from infected tissue) using targeted-sequencing. Analyses indicate a single clonal lineage dominated populations in HA from 2010 to 2016 and targeted-sequencing was useful to estimate ploidy. The implications for adaptation and evolution of P. colocasiae are discussed, as well as consequences for selection and breeding of resistant taro cultivars.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Taro (Colocasia esculenta (L.) Schott) is an important staple root crop with an estimated global production of 10.1 million metric tons in 2014 (FAO 2017). It is widely cultivated in Asia, Africa, South America, Caribbean and the Pacific islands (Kreike et al. 2004; Miyasaka et al. 2013). Taro belongs to the Araceae family and is mainly grown for the starchy corm, although the petiole and leaves, rich in fiber and vitamin C, are also eaten (Huang et al. 2000; Kreike et al. 2004; Miller 1927). Two botanical varieties, based on the shape of the corms, dasheen (var. esculenta) and eddoe (var. antiquorum), have been identified and are thought to be diploid and triploid, respectively (Irwin et al. 1998; Kreike et al. 2004).
Taro leaf blight (TLB) is caused by the oomycete plant pathogen Phytophthora colocasiae Raciborski (Raciborski 1900). Taro leaf blight lowers yield by damaging the photosynthetic area of the leaf and infects the petiole and corm (Brooks 2005). Initially, symptoms appear as small, dark brown flecks on the upper surface of leaves which rapidly expand to become circular, purplish brown to dark brown lesions, often with concentric patterns. Lesions also have typical orange to red brown oozing, with prominent masses of white sporangia surrounding the edge (Brooks 2005; Nelson et al. 2011). Phytophthora colocasiae can produce spores sexually and asexually. The asexual sporangia can directly infect by germinating to produce a germ tube, or indirectly when swimming flagellated zoospores are released in water (Brooks 2005). Phytophthora colocasiae is heterothallic, requiring the interaction of two mating types (A1 and A2) to produce thick-walled sexual oospores (Brooks 2005; Miyasaka et al. 2013; Nelson et al. 2011).
Taro leaf blight is globally distributed and has been found in Asia, Africa, South America, Oceania, Caribbean, and the Pacific territories (http://www.cabi.org/isc/datasheet/40955). For susceptible taro cultivars, yield reduction can be >50% and, in Hawaii, up to 95% leaf reduction has been reported (Nelson et al. 2011). The epidemic caused by TLB in American Samoa in the mid-1990s resulted in dramatic reduction of taro production and decimation of the local susceptible commercial cultivar (Brooks 2005; Miyasaka et al. 2013). Similarly, in 2009, TLB caused a drastic reduction in yield and loss of a susceptible commercial taro cultivar in the Dominican Republic (Miyasaka et al. 2013). Most of the world’s taro production occurs in Africa, and production there decreased from approximately 9.6 million to 6.9 million tonnes between 2008 and 2010 (FAO 2017). This decline in production corresponds with the first reports of TLB occurring in Nigeria and Ghana in 2009 (Bandyopathy et al. 2011; Omane et al. 2012). Nigeria is the world’s leading taro producer, and production during those years fell from 5.4 million tonnes to 2.9 million tonnes. Integrated approaches are used to control P. colocasiae including crop rotation, field sanitation, selection of disease-free vegetative-propagules, pesticides, and TLB-resistant cultivars (Miyasaka et al. 2013; Nelson et al. 2011; Uchida et al. 2002).
The diversity of P. colocasiae has been characterized previously using mating type, proteins, and genetic markers. These include a report of the A1 mating type (n = 144) recovered from taro on the islands of Hawaii, Maui and Kauai and the A2 mating type (n = 799) from Taiwan and, in both cases, the pathogen is presumed to be introduced (Ann et al. 1986; Ko 1979). On Hainan Island, China three mating types (A1, A2, and A0-neuter) were reported, and the authors suggest an Asian origin of P. colocasiae (Zhang et al. 1994). A study of 54 isolates from the Pacific regions, India, and South-east Asia revealed A2 and A0 mating types (Tyson and Fullerton 2007). A recent survey shows that Hawaii and Vietnam have A1 and A2 mating types with A2 dominating more than 95% of isolates. In contrast, A1:A2:A0 mating types from Hainan Island, China, are in the ratio of 69:27:4% (Shrestha et al. 2014). Isozyme and RAPD (Random Amplified Polymorphic DNA) revealed high genetic variation for isolates among and within five countries (Lebot et al. 2003). Mishra et al. (2010) reported unique profiles for 14 isolates analyzed with isozyme and RAPD. Similarly, fine-scale sampling of P. colocasiae from multiple lesions on individual taro leaves in India revealed high levels of genotypic diversity using RAPD and AFLP (Amplified Fragment Length Polymorphism) markers and a surprisingly high level of sequence variation in ITS1 (internal transcribed spacer 1) region, although the authors indicate that the entire ITS1, 5.8S and ITS2 sequence confirms that all isolates are P. colocasiae (Nath et al. 2013a; Nath et al. 2013b). Characterization of populations on the Hawaiian islands, Vietnam and Hainan Island, China, using High Resolution DNA Melting (HR-DM) analysis suggested clonal lineages predominate and some clonal lineages are shared among countries (Shrestha et al. 2014).
Genetic sequencing is providing unprecedented characterization of genetic variation and is useful to measure allele dosage (ploidy) which is difficult to assess using technologies that measure variation indirectly, such as HR-DMA and AFLP (e.g., via fluorescent signals or presence/absence of fragments in a gel matrix). Our previous work using HR-DMA to characterize populations of P. colocasiae revealed instances of Loss of Heterozygosity (LOH) where biological replications (mycelium grown in separate wells but derived from the same isolates) produced heterozygous and homozygous genotypes, similar to what was reported for the closely related vegetable pathogen, P. capsici (Shrestha et al. 2014). How the phenomenon of LOH works is unknown, although recent reports of triploid clonal lineages of P. infestans switching to the diploid state under conditions of stress suggest changes in ploidy may underlie observed LOH and may be part of the evolutionary strategy that makes this group of organisms difficult to study and successful as plant pathogens (Li et al. 2016).
Our initial goal was to assess genome diversity for P. colocasiae recovered from four countries to develop robust SNP markers useful for population analyses. As this work progressed, it became obvious that allele dosage (ploidy) was not homogenous across the P. colocasiae genome and our efforts shifted to focus on the assessment of intra- and inter-genomic variation in ploidy and the implications for genomic instability.
Materials and methods
Sample collection and DNA extraction
For isolates grown in culture, approximately 10-mm sections of taro leaves with typical TLB lesions were excised and placed onto V8-RAP plates (rifampicin 25 ppm, ampicillin 100 ppm, PCNB 25 ppm, 160 mL unfiltered V8 juice, 20 g agar, 3 g calcium carbonate and 840 mL water). A hyphal tip was transferred to V8-RAP agar and, after 3–5 days, a tuft of mycelium was transferred to V8-RAP liquid broth for 5–7 days for mycelium production. Mycelium was lyophilized and genomic DNA extracted using a standard phenol–chloroform extraction method. In addition, genomic DNA was extracted directly from infected tissue. For infected tissue samples, four 7-mm discs were punched from the edge of a distinct lesion using a disposable plastic punch (a section of drinking straw) and then placed into a single well of a 2-ml 96-well plate containing 3–5 3-mm glass beads, freeze-dried, and genomic DNA was extracted as previously described (Lamour and Finley 2006).
Whole genome sequencing and development of de novo reference sequences
Genomic DNA was sheared to 200 bp using a Covaris M220 focused-ultrasonicator (Covaris, Woburn, MA, USA). PCR-free Illumina libraries were built using the KAPA Hyper Prep Kit and the resulting libraries quantified using the KAPA Library Quantification Kit (Kapa Biosystems, Wilmington, MA, USA). Libraries were sequenced at the Oklahoma Medical Research Facility on an Illumina HiSeq3000 device running a 2 × 150 paired-end configuration. The resulting sequences were trimmed based on quality using CLC Genomics Workbench 9.5.2 (CLC-GW) (CLC Bio, Aarhus, Denmark) and processed further to develop P. colocasiae-specific reference contigs and to identify putative SNP sites for population analyses.
A set of nuclear genomic reference sequences for P. colocasiae was developed by de novo assembly of P. colocasiae using CLC-GW at default settings, except only contigs >10Kbp were retained. The resulting contigs were mapped to 18 reference sequences derived from the P. capsici reference genome that contain only those contigs/scaffolds able to be assigned to linkage groups. The P. colocasiae contigs able to be mapped were annotated with open reading frames (ORFs) greater than 300 amino acids using CLC-GW and referred to as the PcoloREF.
Single nucleotide polymorphism (SNP) discovery and target selection
To identify candidate SNP sites, whole genome sequences were mapped to the PcoloREF requiring 90% of a read to have 90% identity, and BAM files exported for further processing using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010). Genotypes were assigned using the diploid HaplotypeCaller followed by hard filtering as recommended by the developers, and custom Perl script was used to extract data for sites with a minimum of ×20 coverage (https://github.com/sandeshsth). Genotypes were assigned as homozygous for alleles at <10% and >90% and heterozygous for alleles between 10 and 90%. A subset of putative SNPs that fall into ORFs and are predicted to be silent were selected for targeted-sequencing and subsequent genotyping. The SNP site in the PcoloREF was changed to an ‘N’ and the flanking sequences extracted as a multi-FASTA file using custom Perl scripts (https://github.com/sandeshsth). Generic primers were designed using BatchPrimer3 v.1.0 (You et al. 2008). Amplification of the target regions and PCR-free library construction (as described above) was conducted by Floodlight Genomics (Knoxville, TN, USA) as part of a no-cost Educational and Research Outreach Program. Each sample had two technical replications and the resulting sample-specific sequence data was made available by FTP transfer. Sample-specific sequences were mapped to the extracted target sequences and processed as above using CLC-GW and GATK.
Genetic analysis
A phylogenetic tree was constructed using all putative silent SNPs across the PcoloREF using the maximum parsimony method with 1000 bootstraps in MEGA7 (Kumar et al. 2016). The initial tree was generated by random addition of sequences (100 replicates) using the Subtree–Pruning–Regrafting (SPR) algorithm with search level 1 (Nei and Kumar 2000). One P. capsici isolate was included as an outgroup. For the isolates and infected plant samples with targeted-sequencing data, samples with identical multi-locus genotypes were identified and a representative genotype retained for further analysis. Allele frequency histograms were constructed using the heterozygous loci from whole genome sequences or targeted sequencing using ggplot2 (Wickham 2009). In addition, for isolates with Whole Genome Sequence (WGS), separate histograms were constructed based on the 18 linkage groups reported for the P. capsici genome.
Results
Isolates
In total, 89 individual isolates of P. colocasiae from Nepal (5), Hawaii (70), China (5) and Vietnam (9) were included; for DNA extraction, mycelium was used for 19 isolates and infected tissue was used for rest of the 70 isolates (Table 1).
Genome sequencing and ploidy
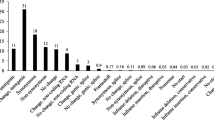
The following seven isolates were sequenced: China (LT8566), Hawaii (LT8771), Nepal (SB4, SB9, and BC13), and Vietnam (LT7290 and LT7291). A total of 42.6 (LT8771), 25.7 (LT8566), 61.7 (LT7290), 39 (LT7291), 14.5 (SB4), 17.92 (SB9) and 24.7 (BC13) million 151-bp paired-end reads were produced and the sequences were deposited in the National Center for Biotechnology Information (NCBI) as BioProject PRJNA378784. The Hawaiian isolate, LT8771, was de novo assembled to produce the PcoloREF. The de novo assembly produced 800 contigs >10Kbp, with an average size of 17,003 bp and N50 of 17,006. A total of 238 contigs (3.8Mbp) mapped to the 18 linkage groups of P. capsici (Table 2). A total of 27,537 putative SNPs were identified in the seven isolates (average of 1 SNP every 138 bp). The proportion of heterozygous loci ranged from 17,894 in the Nepalese isolate, SB9, to 8824 in the Hawaiian isolate, LT8771 (Fig. 1).

Number of homozygous and heterozygous out of 27,537 SNP loci from whole-genome analysis of seven isolates against the de novo reference
Histograms based on the full complement of intragenomic heterozygous allele frequencies revealed distinct (and indistinct) distributions with some isolates appearing to be primarily diploid (Hawaii), triploid (China, Vietnam and Nepal) or some higher level of ploidy (Nepal) (Fig. 2). If ploidy is consistent across the entire genome, distinct modal distributions centering on 50% for diploids, 33 and 66% for triploids, 25, 50 and 75% for tetraploids, etc. are expected. Histograms constructed based on grouping the PcoloREF contigs according to the 18 linkage groups of P. capsici indicate that ploidy is not consistent within a genome (Fig. 3; Supplementary Fig. 1).

Histograms showing intragenomic heterozygous allele frequencies for the heterozygous loci in Fig. 1

Representative intragenomic variation in ploidy for P. colocasiae within potentially linked markers. Data are shown for linkage groups 3, 4 and 5 (based on the linkage groups of P. capsici)
Interestingly, the triploid and higher ploidy isolates had between 14 and 32% more heterozygous loci compared to the diploid isolate from Hawaii. Phylogenetic analysis with 8230 silent (synonymous mutation, does not change the amino acid) SNPs grouped isolates into three clades with isolates from China and Hawaii grouped separately from Vietnam, and these groups being distinct from Nepal where the higher ploidy isolates were more similar than the triploid Nepalese isolate (Fig. 4).

Maximum parsimony tree constructed with 8230 silent SNPs
Targeted sequencing and genotype analyses
In total, 37 SNP markers were assayed in 89 isolates of P. colocasiae from four different countries. The information about the contig, position, and primers of SNP markers is listed in Tables 3 and 4. Multi-locus SNP analysis and clone correction produced seventeen unique genotypes. The genotypes were assigned from G1-G17 (Table 5). Countries did not share genotypes, and Hawaii was dominated by a single clone, G1, with 60 isolates. Although there were many fewer markers, the histograms produced using the heterozygous allele frequencies for the 37 markers provided a reasonable estimate of the predominate ploidy for an isolate (Fig. 5).

Representative histograms showing heterozygous alternate allele frequencies for the 37 SNP loci genotyped by targeted-sequencing for isolates grown in culture (LT) or from infected plant samples (IPS) from Vietnam, Hawaii, China and Nepal
Discussion
Our goal was to develop genomic and genetic resources for P. colocasiae which would be useful for characterizing populations. Once whole-genome and targeted sequencing data were produced; ploidy became the focus of our analyses. It varies within and between countries and varies at different sites within individual isolates. The finding of higher ploidy, especially the triploid state, is not new for the genus Phytophthora, and the late blight pathogen, P. infestans, is often comprised of a few widely dispersed and long-lived triploid clonal lineages. What is new is the detection of intragenomic variations in ploidy for P. colocasiae. We are documenting this same phenomenon, also using WGS and targeted sequencing, of inter- and intra-genomic variability in ploidy for isolates of P. capsici from natural field populations in Taiwan and for single-zoospore isolates from P. capsici recovered from China (unreported data).
It is becoming apparent that the ability to tolerate changes in ploidy may play an important role in the success of asexual lineages for Phytophthora. In P. infestans, most successful clonal lineages are triploid, can persist many years, are dispersed over broad geographical regions, and can vary in ploidy (e.g., triploid reduced to diploid) under stress (e.g., fungicides or starvation) (Li et al. 2016). A recent report on P. capsici isolates recovered from a multi-year, closed field experiment in New York tracked an inbreeding population using >20 K SNP markers produced using the Genotype by Sequencing (GBS) method and also found isolates with increased ploidy, although the exact level of ploidy was difficult to estimate (Carlson et al. 2017). The authors found a mating-type region (MTR) that retained significantly higher levels of heterozygosity, despite inbreeding, and was associated with the A2 mating type. Since P. colocasiae and P. capsici are obligately outcrossing organisms, variable ploidy occurring across a similar MTR may allow P. colocasiae to generate both mating types from single isolates. Taiwan and Vietnam were dominated by a single mating type, whereas Hainan Island had an equal distribution of both mating types (Ann et al. 1986; Shrestha et al. 2014; Zhang et al. 1994).
Previous work indicated that P. colocasiae exists primarily as asexual clones in Hawaii, Vietnam, and China and, in general, country-specific clones dominate, although some clonal lineages were shared between countries (Shrestha et al. 2014). Previous studies also showed that the asexual reproduction is favorable, and that a single mating type, either A1 or A2, dominated the fields (Ann et al. 1986; Ko 1979). The ability to accommodate many intragenomic levels of ploidy provides an additional level of plasticity that may be highly useful for adaptation and surmounting obstacles (e.g., novel resistant hosts or chemicals). During favorable conditions, it could remain at higher ploidy for asexual reproduction and rapid distribution and during adverse conditions, switch to the diploid state to generate both mating types to allow sexual recombination and the production of thick-walled oospores for extended survival outside the plant host (Berman and Hadany 2012; Li et al. 2016). A potentially similar situation occurs under stress in Candida albicans in which concerted chromosome loss in tetraploid zygotes produces diploid strains (Alby and Bennett 2009). Higher ploidy, such as that found in the Nepalese isolates, may explain why some isolates are not able to produce oospores when paired with either A1 or A2 mating types (Shrestha et al. 2014; Tyson and Fullerton 2007; Zhang et al. 1994).
An organism with higher ploidy has a higher preserved variation compared to their diploid counterpart, and this was the case with P. colocasiae where the tetra- or higher ploidy isolates carried more heterozygosity compared to the triploid or diploid isolates. Similarly, triploid P. infestans isolates had higher levels of heterozygosity compared to diploid, including areas of the genome with RXLR and Crinkler (CRN) effector genes (Li et al. 2016). Increased ploidy in genes undergoing positive selection pressures may impact adaptation and vigor, as an increase in beneficial mutations and faster adaptation are reported for tetraploid yeast compared to haploid and diploid counterparts (Selmecki et al. 2015).
Ko suggested that P. colocasiae originated in Asia where there are diverse wild and cultivated taro cultivars (Ko 1979). Interestingly, the isolates from Nepal had higher amounts of intragenomic genetic variation compared to isolates from Hawaii, Vietnam and China. The higher ploidy for isolates from Nepal may be necessary, as taro is widely distributed with different shapes (single-corm, multi-corm, and multi-cormel), and is found in high mountain, hill and plain (terai) regions (Pandey et al. 1998; Rijal et al. 2003). The locally cultivated taro cultivars are diverse and wild-types are common and popular as pig fodder (Pandey et al. 1998). Additional sampling in Nepal will be helpful to better understand the situation.
This study provides important new insight into the kinds of genomic variation possible with P. colocasiae. The potential for intragenomic variation in ploidy, beyond triploid to tetraploid and likely even higher levels, adds a powerful new dimension to the capabilities of P. colocasiae, even if it is existing primarily in the clonal state. Taro leaf blight is an important disease and ongoing work to identify and introgress resistance genes may be impacted by the plasticity of the P. colocasiae genome, especially in cases where single or a few isolates are used to screen promising germplasm. Clearly, much additional work is needed to fully understand the implications for intragenomic variations in ploidy on disease development, evolution, and the development of effective control measures.
References
Alby K, Bennett RJ (2009) Stress-induced phenotypic switching in Candida albicans. Mol Biol Cell 20:3178–3191
Ann P, Kao C, Ko W (1986) Mating-type distribution of Phytophthora colocasiae in Taiwan. Mycopathologia 93:193–194
Bandyopathy R, Sharma K, Onyeka TJ, Aregbesola A, Kumar PL (2011) First report of taro (Colocasia esculenta) leaf blight caused by Phytophthora colocasiae in Nigeria. Plant Dis 95:618. doi:10.1094/PDIS–12–10–0890
Berman J, Hadany L (2012) Does stress induce (para) sex? Implications for Candida albicans evolution. Trends Genet 28:197–203
Brooks F (2005) Taro leaf blight. The Plant Health Instructor. doi:10.1094/PHII2005053101
Carlson MO, Gazave E, Gore MA, Smart CD (2017) Temporal genetic dynamics of an experimental, biparental field population of Phytophthora capsici. Front Genet 8. doi:10.3389/fgene.2017.00026
FAO (2017) FAOSTAT. Available at http://www.fao.org/faostat/en/%23home
Huang A, Titchenal C, Meilleur B (2000) Nutrient composition of taro corms and breadfruit. J Food Compos Anal 13:859–864
Irwin SV, Kaufusi P, Banks K, de la Peña R, Cho JJ (1998) Molecular characterization of taro (Colocasia esculenta) using RAPD markers. Euphytica 99:183. doi:10.1023/a:1018309417762
Ko W (1979) Mating–type distribution of Phytophthora colocasiae on the island of Hawaii. Mycologia 71:434–437
Kreike CM, Van Eck HJ, Lebot V (2004) Genetic diversity of taro, Colocasia esculenta (L.) Schott, in Southeast Asia and the Pacific. Theor App Genet 109:761–768. doi:10.1007/s00122–004–1691–z
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lamour K, Finley L (2006) A strategy for recovering high quality genomic DNA from a large number of Phytophthora isolates. Mycologia 98:514–517
Lebot V, Herail C, Gunua T, Pardales J, Prana M, Thongjiem M, Viet N (2003) Isozyme and RAPD variation among Phytophthora colocasiae isolates from south-east Asia and the Pacific. Plant Pathol 52:303–313
Li Y, Shen H, Zhou Q, Qian K, van der Lee T, Huang S (2016) Changing ploidy as a strategy; the Irish potato famine pathogen shifts ploidy in relation to its sexuality. Mol Plant Microbe In 30:45–52
McKenna A et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next–generation DNA sequencing data. Genome Res 20:1297–1303
Miller CD (1927) Food values of poi, taro, and limu. Bull Bishop Mus 37:1–25
Mishra AK, Sharma K, Misra RS (2010) Isozyme and PCR-based genotyping of epidemic Phytophthora colocasiae associated with taro leaf blight. Arc Phytopathol PFL 43:1367–1380
Miyasaka SC, Lamour K, Shintaku M, Shrestha S, Uchida J (2013) Taro leaf blight caused by Phytophthora colocasiae. In: Lamour K (ed) Phytophthora: a global perspective, 1st edn. CABI, Wallingford, pp 104–112
Nath VS, Senthil M, Hegde VM, Jeeva ML, Misra RS, Veena SS, Raj M (2013a) Genetic diversity of Phytophthora colocasiae isolates in India based on AFLP analysis. Biotech 3:297–305
Nath VS, Senthil M, Hegde VM, Jeeva ML, Misra RS, Veena SS, Raj M (2013b) Molecular evidence supports hypervariability in Phytophthora colocasiae associated with leaf blight of taro. Eur J Plant Pathol 136:483–494
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, Oxford
Nelson S, Brooks F, Teves G (2011) Taro leaf blight in Hawaii. Plant disease bulletin no PD–71, University of Hawaii, Manoa
Omane E, Oduro KA, Cornelius EW, Opoku IY, Akrofi AY, Sharma K, Kumar PL, Bandyopadhyay R (2012) First report of leaf blight of taro (Colocasiae esculenta) caused by Phytophthora colocasiae in Ghana. Plant Dis 96:292. doi:10.1094/PDIS–09–11–0789
Pandey YR, Rijal DK, Sthapit B (1998) Diversity of taro and on-farm conservation through use in Nepal. Ethnobotany and genetic diversity of Asian taro with focus on China. In: Zhu D, Zhou M, Pablo E (eds) Proceedings of the symposium on ethnobotanical and genetic diversity study of taro in China: approaches for the conservation and use of taro genetic resources, 10–12 November, 1998. Laiyang Agricultural College, Laiyang, pp 18–25
Raciborski M (1900) Parasitic algae and fungi, java. Batavia Bull N Y State Mus 19:189
Rijal DK, Sthapit BR, Rana RB, Jarvis DI (2003) Adaptation and uses of taro diversity in agroecosystems of Nepal in: Sthapit B, Upadhyay M, Baniya B, Subedi a, Joshi B (eds) on–farm management of agricultural biodiversity in Nepal. Proceedings of National Workshop, 24–26 April 2001, Lumle, Nepal, pp 29–36
Selmecki AM et al (2015) Polyploidy can drive rapid adaptation in yeast. Nature 519:349–352
Shrestha S, Hu J, Fryxell RT, Mudge J, Lamour K (2014) SNP markers identify widely distributed clonal lineages of Phytophthora colocasiae in Vietnam, Hawaii and Hainan Island, China. Mycologia 106:676–685
Tyson J, Fullerton R (2007) Mating types of Phytophthora colocasiae from the Pacific region, India and south–east Asia. Aust Plant Dis Notes 2:111–112
Uchida JY, Silva JA, Kadooka CY (2002) Improvements in taro culture and reduction in disease levels. Plant disease bulletin no PD–22. University of Hawaii, Manoa
Wickham H (2009) ggplot2: elegant graphics for data analysis. Springer, New York
You FM, Huo N, Gu YQ, Luo M, Ma Y, Hane D, Lazo GR, Dvorak J, Anderson OD (2008) BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9:253. doi:10.1186/1471–2105–9–253
Zhang K, Zheng F, Li Y, Ann P, Ko W (1994) Isolates of Phytophthora colocasiae from Hainan Island in China: evidence suggesting an Asian origin of this species. Mycologia:108–112
Acknowledgements
Permission to harvest infected taro leaves was obtained from all landowners and taro producers, and shipment and processing of isolates was conducted under USDA permit P526P-16-02522 to Kurt Lamour.
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Marco Thines
Electronic supplementary material
Supplementary Fig. 1
Ploidy estimates for P. colocasiae isolates recovered from Hawaii, China, Vietnam and Nepal based on linkage groups defined for the closely related vegetable pathogen, P. capsici. (PDF 204 kb)
Rights and permissions
About this article

Cite this article
Shrestha, S.K., Miyasaka, S.C., Shintaku, M. et al. Phytophthora colocasiae from Vietnam, China, Hawaii and Nepal: intra- and inter-genomic variations in ploidy and a long-lived, diploid Hawaiian lineage.. Mycol Progress 16, 893–904 (2017). https://doi.org/10.1007/s11557-017-1323-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-017-1323-z