
Abstract
The challenges facing potato breeding have actually changed very little over the years with resistance to pests and pathogens remaining high on the agenda together with improvements in storability, reduction in blemishes, and novelty and consistency in cooking/processing qualities. The need to expand the range of targets for potato improvement is being driven by requirements for reduced agrochemical usage and by predictions of the effects of changing climates. Thus fertiliser and water use efficiency are moving up the political agenda. Genetic variation present in germplasm collections needs to be harnessed to provide the genes and alleles required. This paper provides examples of the functional genomics tools and approaches being developed and deployed to provide new options for advancing the breeding of next generation crops. Whilst genetic modification (GM) approaches remain contentious in Europe, this paper will also provide some recent examples of the range of potential impacts that GM approaches could make. It will also consider the value of so-called intragenic or cisgenic approaches to potato genetic engineering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Functional Genomics has been defined by some as “The Force Behind the Future of Plant Breeding” and can be simply defined as approaches deployed to identify genes and their associated functions. It clearly complements the more structural aspects of genomics which includes genome sequencing, the chromosomal mapping of genes to specific traits and association between phenotype and structural diversity within alleles. The challenge for functional genomics lies not only in ascribing the functions of individual genes but also in analysing the potential interactions between genes and gene families in determining, both temporally and spatially, the development of target traits. It follows that to deliver on functional genomics we require the appropriate experimental tools which include high-throughput gene expression analysis (transcriptomics), bioinformatics platforms, contemporary analytical (e.g. metabolomics, proteomics) and phenotyping platforms and the capacity to test functionality using both forward and reverse genetic approaches (e.g. transgenics, mutants).
Functional Genomics: Targets for Potato Improvement
The EU project EUROCROP (www.eurocrop.cetiom.fr) aims to define a common vision for the future of research and development related to arable crops in Europe and includes potato as one the crop species. EUROCROP brings together concerned stakeholders and actors, to reach a collective analysis of research needs in order to improve the European arable crops competitiveness, and propose appropriate action. In its deliberations to date the project has identified several areas for a prioritised research focus which can benefit from advances in genomic and functional genomic approaches. The suggested targets reflect the need to maintain and grow the markets but also to address generic issues such as human health and nutrition, the impact of climate changes, EU regulatory and legislative requirements, and the need to develop sustainable and profitable production systems with reduced inputs (and cost). Some of the priority areas identified which functional genomics can contribute to (within the framework of plant breeding) include:
-
1.
Improving utilisation of potato in processed products: e.g. develop processed products and convenience foods with improved taste and texture. Exploit new cooking/processing technologies. Develop potato-based products with low Glycaemic Index.
-
2.
Addressing consumer demands for healthy food: e.g. develop tools to design better diets for individuals and an improved understanding of the role of compounds in potatoes that can contribute to healthy diets.
-
3.
Improving resource use efficiency: e.g. develop germplasm tolerant to drought, higher CO2 levels, and which can maintain yield and quality with reduced nutrient inputs. Assess the potential impact of climate change on quality.
-
4.
Improving pest and disease resistance and knowledge of pathogen evolution: e.g. exploit information from genome sequencing initiatives to develop germplasm with multiple trait improvements.
“Omics” Technologies as Components of the Functional Genomics Toolkit
The ‘Central Dogma Theory of Genetics’ states that DNA is transcribed into RNA that is then translated into protein. Proteins are intrinsic in the establishment and maintenance of biochemical pathways in an organism that lead to the production/turnover of various metabolites. Thus the different “omic” technologies are inter-related in that: transcriptomics aims to assess changes in the transcriptome (the entire complement of RNA produced by DNA transcription of a cell, tissue or organism at a particular time point), proteomics studies the total protein complement (the proteome), and metabolomics the complement of small molecules (low molecular weight, <1,500 Da). Depending on the species, plants contain ca 30,000 to 60,000 genes, with an estimated 100,000–120,000 proteins (this could be a significant underestimate) and, across the plant kingdom, ca 90,000 to 200,000 metabolites. However, the number of metabolites in a single species will be much smaller.
Microarray technology is the most common approach used for gene expression profiling. Microarrays make use of the information created by genome sequencing (more than 600 complete genomes sequenced to date [www.genomesonline.org]) and from the myriad of expressed sequence tags (ESTs) which provide information on genes expressed in specific cells, tissues organs at a particular time and under particular conditions. The microarray technologies will not be described in detail but gene expression technologies have greatly matured over the past years, and it has become clear that hybridisation-based approaches have obvious limitations in cross-species comparisons (Gilad et al. 2006). However, sequencing-based approaches could be used to quantify gene expression if the sequence reads could be unambiguously mapped to the corresponding transcripts. While the short sequence reads of serial analysis of gene expression (SAGE) and related techniques are severely limited by the requirement of a reliable genome annotation, the recently developed 454 sequencing technology (Margulies et al. 2005) may provide sufficient sequence information to overcome this limitation at moderate costs. Furthermore, due to ability to identify Single Nucleotide Polymorphisms (SNPs) in transcripts, the method may provide a powerful solution for measuring allele-specific gene expression.
Potato Transcriptomics
Most potato microarray experiments performed to date have used the widely accessible spotted cDNA array produced by The Institute for Genomic Research (TIGR) which contains around 10,000 cDNA clones (http://www.tigr.org/tdb/potato/microarray_desc.shtml). This has proven to be a useful resource and has been applied to a wide range of potato developmental issues. A particularly useful feature is that many of the results using this array are catalogued at http://www.tigr.org/tdb/potato/microarray_desc.shtml. Other groups have developed their own arrays specifically focused on particular aspects. For example Kloosterman et al. (2005) developed a cDNA array to describe changes in gene expression during tuber development. Using this array, 1,315 genes were identified that were strongly differentially expressed during tuber development. However, the recently developed Potato Oligo Consortium (POCI) array containing 44,000 features is the best microarray platform currently available to analyse global gene expression in potato (described in Kloosterman et al. 2008) representing 42,034 unigene sequences, thereby enabling a much more complete analysis of gene expression than has hitherto been achievable. It has been estimated that 35,000 genes are expressed in tomato (Van der Hoeven et al. 2002) and a similar number are probably expressed in potato, although it has been suggested that the number of expressed genes in potato may be somewhat higher (Datema et al. 2008). Therefore the TIGR array probably only contains ca. 35% of the transcriptome, which is a serious limitation for global expression studies. Approximately 50% of the sequences on the POCI microarray were not represented on the TIGR array (Kloosterman et al. 2008). The initial report by Kloosterman et al. (2008), demonstrates the utility of the POCI microarray and it is anticipated that the array will be applied to help identify key genes in many aspects of tuber quality and development. Some of these experiments are underway within the EU-SOL project (http://www.eu-sol.net/public). Additionally, the array is being used to address the regulation of dry matter accumulation, sugar accumulation and after-cooking darkening by a Canadian consortium (Regan et al. 2006).
Potato Proteomics
Lehesranta et al. (2005) assessed potato tuber proteome diversity using a range of germplasm. In addition, a selection of genetically modified (GM) potato lines was compared to assess the potential for unintended differences in protein profiles. Clear qualitative and quantitative differences were found in the protein patterns of the varieties and landraces examined, with 1,077 of the 1,111 protein spots analysed showing statistically significant differences. The diploid species Solanum phureja could be clearly differentiated from tetraploid (Solanum tuberosum) genotypes. Many of the proteins apparently contributing to genotype differentiation were involved in disease and defence responses, the glycolytic pathway, sugar metabolism or protein targeting/storage. Only nine proteins out of 730 showed significant differences between GM lines and their controls. There was much less variation between GM lines and their non-GM controls compared with that found between different varieties and landraces. With regard to the impact of crop production practices, Lehesranta et al. (2007) compared proteomes of organic and conventionally grown potato and showed that fertility management practices had a significant effect on protein composition. Quantitative differences were detected in 160 of the 1100 tuber proteins separated by 2D-gel electrophoresis. Proteins identified were involved in protein synthesis and turnover, carbon and energy metabolism and defence responses, suggesting that organic fertilisation leads to an increased stress response in potato tubers.
Potato Metabolomics
Studies have shown that there is not necessarily a good and direct relationship between gene expression and relative levels of metabolites. This is understandable as metabolite levels can be determined, for example, both by the activities of enzymes in the pathway concerned and by fine-control mechanisms involving enzyme effectors, co-factors, etc. Urbanczyk-Wochniak et al. (2003) showed that specific transgenic potato lines could not be separated either from each other or from the wildtype using transcript profiling (even though the transgenes produced a large effect on tuber metabolism). However, metabolite profiling revealed that transgenics clustered completely independently of each other and of the wild type control, indicating that metabolite pool sizes can be a more sensitive indicator of phenotype than transcript levels and hence potentially a more powerful phenotyping technology. The authors also showed that combined analysis of transcript and metabolite profiling by pairwise correlation analysis indicated that only 25% to 36% of the variation in metabolite level was attributable to variation in transcript level. Some other novel correlations were revealed that could stimulate the exploration of unsuspected regulatory circuits. It is also important to point out that metabolites change according to developmental, physiological, and plant “health” status, thus biological variance in metabolite levels can be very large. Also, metabolites are structurally diverse chemically so no single extraction or analytical technique is suitable for all low molecular weight metabolites.
Defernez et al. (2004) compared the metabolomes of non-GM and some specific GM potato tubers with modified development and/or metabolism. Despite the fact that many of the GM plants included in the study were phenotypically very different from their non-GM parents, and that several hundred compounds could be compared, the number of differences that could be found between GM and non-GM tubers was extremely small. Similarly, Catchpole et al. (2005) described a comprehensive comparison of tuber metabolites in field-grown GM potato (genes inserted to induce fructan biosynthesis) and conventional potato tubers using a hierarchical approach. Their data also showed that, apart from targeted changes, the GM potatoes were equivalent to the traditional non-GM comparators.
Challenges for Data Analysis
“Omics” outputs are extremely data rich thus data mining and reliable comparative analysis are central requirements. Despite the ever-expanding range of technological options, significant challenges remain that relate to the acquisition, interpretation and statistical treatments of “omics” data (see Broadhurst and Kell 2006; Hall 2006). As pointed out by Scholz et al. (2004), the development of software tools to enable in-depth analysis of any list of interrelated biological data (pathway analysis tools) is evolving. Several metabolic pathway databases are available to facilitate our understanding of transcriptome and metabolome data. For example, The Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.ad.jp/kegg/) has a pathway database (PATHWAY) that contains information on metabolites and genes, as well as graphical representations of metabolic pathways and complexes derived from various biological processes.
Systems Approaches
Whilst there are many potential uses for gene expression arrays, it is not simply mRNA levels that need to be considered, but also the amount and modification of proteins expressed that determine true gene activity. An important goal is therefore to couple transcriptomic data to the outputs from the other “omics” tools, proteomics and metabolomics to develop a truly integrated understanding of biological processes which regulate crop plant composition, for example. These “omics” tools in turn must be linked to DNA sequences and sequence variation to better understand the processes which contribute to biological variation. The goal of “omic” approaches is therefore to acquire a comprehensive, integrated understanding of biology by studying all biological processes to identify the various players (e.g., genes, RNA, proteins and metabolites) rather than each of those individually. This systems biology approach is highly challenging.
Transgenic Potato—Progress in Trait Modification and Gene Functionality Testing
Commercialisation Issues
Potato continues to be an important experimental system for the functionality testing of genes using transgenic approaches. Examples will be provided later in this overview.
It is worth pointing out, however, that on the global map of commercial transgenic crop production, potato hardly figures. Since the demise of the Monsanto Company’s NatureMark unit and the removal of its virus and insect resistance products from the market no new commercial GM products have been launched. However, the first GM potato to be cultivated in Europe is likely to be Amflora, a pure amylopectin starch potato developed by BASF Plant Science by switching off the gene for the Granule Bound Starch Synthase (GBSS), the key enzyme for the synthesis of amylose. Amflora is designed for use in technical, non food applications. Whilst is has been assessed as safe by the European Food Safety Authority, it has not yet been approved for commercial production as concerns have been raised over the fact that the potato contains the nptII gene marker gene which confers resistance to specific antibiotics. Thus, despite the fact that major, multitrait improvements to this genetically complex, vegetatively propagated crop, can almost certainly only come from transgenic approaches, progress in commercialisation remains stagnant. Indeed, even the numbers of GM field trials for experimental purposes have dwindled.
Not withstanding the political challenges faced, research must continue to develop approaches that take us beyond the current state-of-the-art with regard to (a) the need to develop more efficient GM breeding approaches, (b) the need to develop efficient approaches for stacking or pyramiding genes to deliver multiple improvements in crop utility and performance traits. For tetraploid potato transgene stacking through traditional crossing is not a practical solution. Also, the costs of the chain of processes involved in GM commercialisation can be high (e.g., ca $6 to 15 million for herbicide tolerant or insect tolerant maize (Kalaitzandonakes et al. 2007)). Since, within the EU each transgenic event needs to be individually risk assessed, on most occasions it will be more cost effective to target multitrait modification in a single transformation event or via co-transformation. Such approaches also make sense in terms of durable disease resistance as the deployment of several gene targets should help delay resistance breakdown. The development of co-transformation strategies, improved transformation efficiencies for recalcitrant potato sub-species (Ducreux et al. 2005b) and an increasing range of potato promoters are all helping to advance the technology for potato.
Cisgenic/Intragenic Approaches—A Way Forward?
One new method that combines the benefits of traditional breeding and genetic engineering, but circumvents many of their issues, is represented by the intragenic approach (Rommens et al. 2007 and references therein). It isolates specific genetic elements from a plant, recombines them in vitro, and inserts the resulting expression cassettes into a plant that belongs to the same sexual compatibility group using plant-derived transfer DNAs (P-DNAs) and marker-free transformation. The intragenic approach has been used to produce a quality-enhanced potato with reduced ‘black spot bruise,’ and lower reducing sugars out of cold storage (improved processing quality), reduced amounts of processing-induced acrylamide, and increased starch levels (Rommens et al. 2006). In a cisgenic approach a recipient plant is also modified with a natural gene from a sexually compatible plant. However, in this case the gene includes its native introns and is flanked by its native promoter and terminator in the normal sense orientation. The argument is that in a cisgenic plant, the gene of interest, together with its promoter, has already been present in the species or in a sexually compatible relative for centuries. Therefore cisgenesis does not alter the gene pool of the recipient species and provides no additional traits (Schouten et al. 2006).
The annotated genome sequence of potato will be available in the not too distant future and will facilitate comprehensive searching in germplasm collections for novel alleles that represent variant versions of genes, or genes with altered functions. This will provide a source of DNA sequences for transfer via genetic engineering approaches from within the gene pools already utilised by plant breeders. This would assist the intragenic/cisgenic approach and provide, for example, opportunities for targeted genetic changes in biochemical pathways to accumulation of specific metabolites for specific functions or to transfer disease resistance genes from related wild species. The transfer of genes between plants of the same or closely related species does not seem to raise similar ethical concerns in the GM debate as transfer of genes from unrelated species.
High Throughput Analysis of Gene Function, Mutants and Activation Tagging
The generation of stable transgenic/intragenic lines is a very powerful tool in functional genomics. However a limitation is that the creation of transgenic lines is both time consuming and resource intense. A higher throughput approach to determine gene function is the use of virus induced gene silencing (VIGS). For example a vector based on potato virus X (PVX) is effective in triggering a VIGS response in both diploid and tetraploid potato (Faivre-Rampant et al. 2004c). The response is most useful for leaf phenotypes although systemic silencing of a phytoene synthase gene was observed in potato tubers. However, attempts to develop this approach into a high throughput screen for a wider range of genes in tubers has demonstrated that the effects on tuber gene expression can be inconsistent and further developments are required to make this a robust approach (Lacomme and Taylor unpublished). VIGS vectors based on tobacco rattle virus have also proved effective in leaves from a range of Solanum species including potato (Brigneti et al. 2004).
In contrast to the situation in Arabidopsis there are very few potato mutants available in potato. A recent breakthrough is the development of a potato diploid TILLING population (Muth et al. 2008). This is the first report of rapid and efficient mutation analysis and breeding in potato. Although initially focusing on starch biosynthesis as a well-characterized model, a TILLING approach could be applied to any desirable trait, a goal that will become more defined as information emerges from the potato genome sequencing project (www.potatogenome.net) and functional genomics initiatives (Gebhardt 2004). By combining new sequence and functional information with a TILLING approach, it will be possible to generate and integrate new source alleles rapidly into elite breeding cultivars, an approach that has not been possible previously.
The potential to form mutants in complex plant genomes has also increased recently with the development of activation tagging which results in the generation of dominant gain-of-function mutants. In this approach Agrobacterium-mediated transformation is used to introduce a strong promoter such as the Cauliflower Mosaic virus 35S promoter, randomly into the genome (Weigel et al. 2000). When the promoter inserts adjacent to a gene, over-expression can result. Transgenic lines can be selected for a phenotype of interest and the over-expressed gene can be identified using PCR-based approaches. So far the Canadian potato genome project has generated over 8,000 mutant lines and these are likely to be a valuable resource (http://www.cpgp.ca/Resources/plants/description.php).
Potato Functional Genomics and Quality Traits
Potato consumption faces significant competition from other staple sources of carbohydrates such as rice and pasta. Strategically, therefore, the development of well differentiated potato products is an important goal. Tuber quality traits such as flavour, texture and nutritional value are assuming a greater importance in R&D since consumers are becoming increasingly demanding with respect to convenience, quality, novelty, eating experience, nutritional value and safety (McGregor 2007). Additionally, it is recognized that tuber quality is closely associated with tuber life cycle issues. For example the control of tuber initiation impacts on crop scheduling, yield and uniformity (Vreugdenhil and Struik 1989; Struik et al. 1990; Struik et al. 1991; Ewing and Struik 1992; Fernie and Willmitzer 2001; Hannapel 1991; Jackson 1999; Rodriguez-Falcon et al. 2006) and the control of tuber dormancy is critical in tuber storage (Suttle 2004). Thus a major challenge is to develop market advantage by producing cultivars and processed products that exhibit distinctive and desirable characteristics. However, as with many food crops, quality and nutritional/developmental traits are probably driven by multi-factorial parameters and hence are difficult to assess in breeding programmes. In recent years functional genomics tools to understand key quality traits have been developed and this report will review some of these approaches.
Comparative Gene Expression Profiling of Potato Germplasm
The gene pool used to develop cultivated potatoes is considered to be fairly narrow and has not widely utilized the full diversity (and hence characteristics) of potato germplasm available. Efforts to broaden the genetic base of cultivated potatoes have been made, with some success (Bradshaw and Ramsay 2005). A valuable resource in this regard is S. tuberosum group Phureja, differentiated from S. tuberosum group Tuberosum on the basis of a number of important tuber quality traits such as flavour, texture, colour and reduced tuber dormancy (De Maine et al. 1993, 1998; Dobson et al. 2004; Morris et al. 2004; Ghislain et al. 2006). Despite being able to differentiate the Phureja group of landraces based on geographical origin, this group is very similar genetically to the Tuberosum group (Spooner et al. 2007). Whatever the final taxonomic outcome, as many of the Phureja group can be clearly differentiated based on tuber quality trait parameters, they form a useful resource for the identification of key genes that underpin these traits.
In view of the Phureja/Tuberosum trait differences, comparison of metabolite and transcript profiles in tubers from Phureja and Tuberosum cultivars has been carried out in order to gain insight into the traits of interest (Shepherd et al. 2007; Ducreux et al. 2008 submitted). For example, using the POCI array, gene expression was compared in tubers from two Solanum tuberosum group Phureja cultivars and two S. tuberosum group Tuberosum cultivars. Three hundred and nine genes were significantly and consistently up-regulated in Phureja whereas 555 genes were down-regulated (Ducreux et al. 2008 submitted). Approximately 46% of the genes in these lists can be identified from their annotation and amongst these are candidates that may underpin the Phureja/Tuberosum trait differences. For example, a sesquiterpene synthase gene was identified as being more highly expressed in Phureja tubers and its corresponding full length cDNA was demonstrated to encode α-copaene synthase. Other potential “flavour genes”, identified from their differential expression profiles, include a gene encoding branched-chain amino acid aminotransferase. Major differences in the expression levels of genes involved in cell wall biosynthesis (and potentially texture) were also identified including genes encoding pectin acetylesterase, xyloglucan endotransglycosylase and pectin methylesterase.
Transgenic Approaches to Quality Improvement—Examples
Potato was one of the first plants to be genetically transformed (Ooms et al. 1983) and the approach remains important for proof of gene function today. In relation to tuber carbohydrate metabolism, transgenic biology has played an important role in developing our understanding of the processes involved in sucrose-starch inter conversion (reviewed in Hofius and Bornke 2007). Accumulation of dry matter remains an important quality trait and as well as helping to define the role of key enzymes in starch biosynthesis, transgenic biology has demonstrated that notable improvements in starch accumulation are possible. A recent example from Zhang et al. (2008) demonstrates how carbon and energy are co-limiting for starch synthesis. Simultaneous over-expression of two genes, a glucose-6-phosphate/phosphate translocator and an adenylate translocator, results in an increase in starch content of 28%. Interestingly, over-expression of either gene alone resulted in no increase, giving valuable insights into the limiting factors for starch biosynthesis. Cold sweetening of tubers in storage is another important quality issue where transgenic approaches have proved interesting (for example Greiner et al. 1999). However, as more than 80 proteins are known to be involved in carbohydrate metabolism, profiling of metabolites and transcripts will be required to give a more complete understanding of the subtle interplay of gene expression and metabolite levels. Nevertheless, Rommens et al. (2006) have shown, using an intragenic approach, that sufficient changes in tuber carbohydrate can be engineered to develop commercially viable products.
Other tuber traits have also benefited from transgenic approaches. For example transgenic manipulations of the carotenoid biosynthetic pathway in potato tubers have been carried out at several different laboratories. Firstly it was demonstrated by Romer et al. (2002) that zeaxanthin epoxidase down-regulation resulted in a marked tuber carotenoid phenotype. In some transgenic lines, total carotenoid content was increased up to six-fold with a particularly high accumulation of zeaxanthin. Expression of a bacterial phytoene synthase gene in potato resulted in a similar increase in total carotenoid content, although in this case the profile of individual carotenoids was quite different, with β-carotene, lutein and violaxanthin dominating the carotenoid profiles (Ducreux et al. 2005a). More recently even higher levels of tuber carotenoids have been achieved transgenically by co-expressing several of the pathway genes, resulting in potato tubers that accumulate up to 60 μg/g dry weight β-carotene, significantly higher than the best levels achieved in “Golden Rice” (Diretto et al. 2007). Although the transgenic experiments clearly demonstrate the metabolic flexibility of potato tubers in this regard, the regulators of carotenoid metabolism in yellow-fleshed germplasm remain to be fully understood. Genetic approaches have enabled some progress, resulting in the identification of an allele of β-carotene hydroxylase as a candidate gene for the Y-locus, a major QTL for carotenoid content in some potato germplasm (Brown et al. 2006). As the drive for more nutrient dense stable food intensifies, it is likely that transgenic enhancement of potato micronutrients will become more prevalent. Targets are likely to include B and C type vitamins and mineral content such as potassium and iron.
Potato Functional Genomics and the Tuber Life Cycle
Tuberisation
As well as metabolic traits, tuber development also impacts heavily on tuber quality. In recent years it has become apparent that the photoperiodic control of flowering and tuberisation share elements with similar functions (Rodriguez-Falcon et al. 2006). For example, the photoperiodic control of tuberisation requires phytochrome B (Jackson et al. 1996). Homologues of GIGANTEA, CONSTANS and flowering locus T, elements well characterized in the day length control of flowering pathways of Arabidopsis and rice (Koornneef et al. 1991), are also implicated in the short-day pathway controlling tuberisation in potato (Rodriguez-Falcon et al. 2006). For example in Arabidopsis, expression of the transcription factor CONSTANS accelerates flowering in response to long days (Putterill et al. 1995). Constitutive over-expression of the Arabidopsis CONSTANS gene in potato results in a delayed tuberisation phenotype (Martinez-Garcia et al. 2002).
Plant growth regulators have also been implicated in the control of tuber initiation. In particular the role of gibberellins in this respect has become clearer in recent years. It has been demonstrated that there is a decrease in the levels of GA1 in the stolon tip at the onset of tuberisation of microtubers grown in vitro, resulting in the re-orientation of microtubules and a shift in the plane of cell division. The regulation of GA levels may occur via transcriptional control (StBEL5, POTH1; Chen et al. 2004) or regulation may be brought about by altered sensitivity to GA (PHOR1; Amador et al. 2001). Transgenically manipulated changes in the expression levels of these genes affect the onset of tuberisation. More recently, the up-regulation of a GA2-oxidase gene early in the tuber initiation process, in the sub-apical region of the stolon provides another mechanism whereby the GA level in this region is decreased (Kloosterman et al. 2007). Thus it appears that there are two main pathways controlling the onset of tuberisation—a short day (or more accurately a long night) dependent pathway and a GA dependent pathway. It remains to be clarified how these two pathways interact. A genetic approach may provide the way forward, as already 11 quantitative trait loci (QTL) affecting tuberisation have been defined in reciprocal backcrosses between Solanum tuberosum and S. berthaultii (van den Berg et al. 1996). With the potato genome project underway and the development of high throughput gene mapping platforms the identification of genes underlying these QTL is likely to occur in the next few years. It may then be possible to understand at the molecular level, the adaptive changes that occurred in modern germplasm as the response to day length has been selectively modified. Furthermore, the goal of producing a crop that is more coordinated in tuber initiation may finally be attainable.
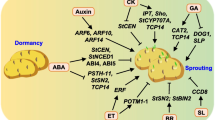
Dormancy
Functional genomics approaches are currently being explored to enhance our understanding of the control of tuber dormancy. For example, it appears that the potato tuber life cycle is controlled by cycles of meristem activation and deactivation, mediated via symplastic association and disassociation of the tuber apical bud (Viola et al. 2001a). Thus on dormancy release, the apical bud regains symplastic connection with the tuber and growth resumes (Viola et al. 2001b). Subsequent work examining dormancy release in buds of the mature tuber identified molecular markers in potato tuber buds that were induced or repressed specifically on release from endodormancy, in some cases prior to any visible sign of growth (Faivre-Rampant et al. 2004a, b).
Other studies have demonstrated that potato tuber sprouting can be controlled by manipulation of carbohydrate metabolism (Geigenberger et al. 1998; Sonnewald 2001; Hajirezaei et al. 2003). Tuber sprout growth is initially supported by energy captured from sucrose breakdown. As inorganic pyrophosphate is a necessary co-factor for sucrose breakdown, removal of pyrophosphate by expression of a bacterial pyrophosphatase in transgenic tubers increases sucrose content and prevents its use as an energy supply. Consequently, sprout growth is significantly inhibited when sucrose is limited but accelerated when sucrose supply is increased. Transgenic approaches have also started to address the role of the plastid-derived isoprenoid hormones (particularly, cytokinins, gibberellins and abscisic acid) in the control of potato tuber dormancy. For example, over-expression of the gene encoding the first step of the isoprenoid biosynthetic pathway (catalysed by 1-deoxy-d-xylulose 5-phosphate synthase) leads to a much clearer separation of dormancy release and sprouting, enabling the initiating events in dormancy release to be temporally isolated from subsequent sprout growth (Morris et al. 2006). As for tuber initiation, the use of microarray analyses, transgenic and genetic approaches, will enable the identification of the key regulatory genes in the coming years.
Functional Genomics—Pest and Disease Resistance
There are now several examples of the isolation of genes (R genes) conferring resistance to the major biotic threats to potato, such as viruses, cyst nematodes (Globodera rostochiensis and G. pallida), and late blight, caused by the oomycete Phytophthora infestans. The majority of successful R gene isolation projects have used a map-based approach, involving the construction of dense genetic maps and large-insert genomic libraries. All potato R genes isolated to date have conformed to the nucleotide binding site-leucine rich repeat (NBS-LRR) type. The first potato R gene isolated by map based means was the PVX resistance gene Rx1, introgressed into cultivated potato from the primitive cultivated species S. tuberosum ssp. andigena accession (Bendahmane et al. 1999). The Rx2 gene on chromosome V was cloned by simple homology to Rx1 (Bendahmane et al. 2000). The isolation of Rx1 aided considerably the isolation of the nematode resistance gene Gpa2, which resides with Rx1 in a resistance gene ‘hotspot’ on chromosome XII (van der Vossen et al. 2000), containing four very closely related resistance gene ‘homologues’. The work on the Gpa2/Rx1 ‘cluster’ has yielded important information concerning the evolution and structure of R gene loci and has shown beyond any doubt that resistances to different pests and pathogens can be coded by structurally similar genes from the same ‘local’ cluster. This work also led to the deposition of the first large genomic fragment sequence (∼186 kb) into the genomic databases, and to one of the first uses of a PCR based approach for the ‘mining’ of R gene candidates from potato (Bakker et al. 2003). This strategy has the potential for providing significant short-cuts in the isolation of further functional R genes.
Resistance to the oomycete pathogen P. infestans is conferred by a large and ever increasing number of R genes, the first to be isolated being the R1 gene (Ballvora et al. 2002). R1 is a member of the leucine zipper/NBS/LRR class of plant resistance genes, and is similar to the Prf gene, which confers resistance to Pseudomonas syringae in tomato. R1 maps to a hotspot for resistance in the GP21–GP179 region of chromosome V and was introgressed into cultivated potato from the wild hexaploid Mexican species S. demissum. On Agrobacterium-mediated transformation with a BAC subclone harbouring the candidate resistance gene, nine out of twelve transgenic lines tested exhibited the resistance phenotype against the appropriate P. infestans race. The resistance allele was observed to represent an insertion of a functional resistance gene with respect to the susceptible allele contained in the heterozygous resistant parent. An interesting and novel development of transgenic technology was the use of a biolistic approach with large DNA fragments to achieve stable transformation and functional complementation of the susceptible phenotype with a BAC clone (BA87d17), containing the R1 gene (Ercolano et al. 2004). Thirty-one kanamycin resistant plants were regenerated of which thirteen showed the necrotic lesions typical for the hypersensitive response after infection with the incompatible P. infestans race 4, which carries the avirulence gene Avr1. As with Gpa2/Rx the work of various groups in isolating and sequencing BAC clones from the R1 region has yielded a great deal of information concerning the structure of the potato genome and of resistance hot-spots. A notable example is the work of Kuang et al. (2005), who completely sequenced a large number of BAC clones from the R1 region of the allohexaploid S. demissum. These authors report extremely large amounts of divergence, both in length and gene content, among the three homoeologous haplotypes. This and other related work (e.g. Ballvora et al. 2002) provides significant insights into the evolutionary history of a resistance hotspot in potato, and which will no doubt help in the isolation of other resistance genes and QTLs from the same region.
Cloning of the Gro1-4 gene, which confers resistance to pathotype Ro1 of the cyst nematode G. rostochiensis has been achieved using a candidate gene approach (Paal et al. 2004). The gene was co-localised in a large segregating population with a sequenced AFLP marker derived from a ‘resistance-gene-like’ sequence. The marker was used to isolate 15 members of a closely related gene family from genomic libraries. By taking into account different types of information (inheritance patterns in resistant and susceptible germplasm, mapping data, DNA sequence information) it was possible to reduce the number of candidates to three genes, which were subsequently tested for complementation of a susceptible phenotype by stable transformation. The identified functional gene, a member of the TIR-NBS-LRR class, differs from susceptible members of the same family by 29 amino acid changes. This study may pave the way for other resistance gene cloning efforts in potato using a similar approach. It is hoped that this type of approach may also lead to the isolation of QTLs conferring partial and relatively durable resistance to the major potato pests and pathogens, which to now has remained an elusive goal. Such a target QTL is that reported by Bradshaw et al. (2006), which maps on chromosome IV close to several major R genes which may be cloned in the near future (Park et al. 2005).
Since these groundbreaking resistance gene isolation efforts, several other R genes, too numerous to describe here, have been isolated from potato. Moreover, there are ongoing attempts to clone several others, notably as part of the BIOEXPLOIT project, funded by the EU (http://www.bioexploit.net/). R gene isolation is still heavily dependent on extensive genetic mapping allied to a transgenic approach for the confirmation of function, although now that most of the R gene ‘hot spots’ have been detected and characterised, at least partially, at the sequence level, the ability to ‘mine’ candidate resistance genes from a locus has increased greatly. What now remains is to develop the tools for rapidly monitoring the function of the large numbers of candidate alleles detected in PCR-based screens. As mentioned previously in this article Brigneti et al. (2004) used a TRV vector to silence various potato R genes (e.g. R1, Rx, RB) generating susceptible phenotypes in ‘detached leaf’ assays. However, the likelihood that VIGS will lead to silencing of entire families of closely related R genes suggests that it will not provide a definite positive indication of the functionality of a particular candidate R gene sequence. A highly specific ‘gain of function’ assay, not requiring stable transformation, is still very much the ‘holy grail’ of potato resistance genetics. Recent advances in pathogen ‘effectoromics’ (Kamoun 2007) may ultimately pave the way, whereby pathogen free systems able to detecting specific recognition of pathogen-secreted effector molecules by individual cognate R genes will identify functional variants for testing via transgenic routes.
References
Amador V, Bou J, Martinez-Garcia J et al (2001) Regulation of potato tuberisation by day length and gibberellins. Int J Dev Biol 45:S37–S38
Bakker E, Butterbach P, Rouppe van der Voort J et al (2003) Genetic and physical mapping of homologues of the virus resistance gene Rx1 and the cyst nematode resistance gene Gpa2 in potato. Theor Appl Genet 106:1524–1531
Ballvora A, Ercolano MR, Weiss J et al (2002) The R1 gene for potato resistance to late blight (Phytophthora infestans) belongs to the leucine zipper/NBS/LRR class of plant resistance genes. Plant J 30:361–371
Bendahmane A, Kanyuka K, Baulcombe DC (1999) The Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11:781–791
Bendahmane A, Querci M, Kanyuka K (2000) Agrobacterium transient expression system as a tool for the isolation of disease resistance genes: application to the Rx2 locus in potato. Plant J 21:73–81
Bradshaw JE, Ramsay G (2005) Utilisation of the commonwealth potato collection in potato breeding. Euphytica 146:9–19
Bradshaw JE, Hackett CA, Lowe R et al (2006) Detection of a quantitative trait locus for both foliage and tuber resistance to late blight [Phytophthora infestans (Mont.) de Bary] on chromosome 4 of a dihaploid potato clone (Solanum tuberosum subsp tuberosum). Theor Appl Genet 113:943–951
Brigneti G, Martin-Hernandez AM, Jin HL et al (2004) Virus-induced gene silencing in Solanum species. Plant J 39:264–272
Broadhurst DI, Kell DB (2006) Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2:171–196
Brown CR, Kim TS, Ganga Z et al (2006) Segregation of total carotenoid in high level potato germplasm and its relationship to beta-carotene hydroxylase. Am J Potato Res 83:365–372
Catchpole GS, Beckmann M, Enot DP et al (2005) Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proc Natl Acad Sci USA 102:14458–14462
Chen H, Banerjee AK, Hannapel DJ (2004) The tandem complex of BEL and KNOX partners is required for transcriptional repression of ga20ox1. Plant J 38:276–284
Datema E, Mueller LA, Buels R, Giovannoni JJ, Visser RGF, Stiekema WJ, van Ham RCHJ (2008) Comparative BAC end sequence analysis of tomato and potato reveals overrepresentation of specific gene families in potato. BMC Plant Biol 8:34. doi:10.1186/1471-2229-8-34
Defernez M, Gunning YM, Parr AJ et al (2004) NMR and HPLC-UV profiling of potatoes with genetic modifications to metabolic pathways. J Agric Food Chem 52:6075–6085
De Maine M, Carrol CP, Torrance CJW (1993) Culinary quality of tubers derived from Solanum phureja and Solanum tuberosum × Solanum phureja hybrids. J Agric Sci 120:213–217
De Maine M, Lees A, Bradshaw J (1998) Soft-rot resistance combined with other tuber characters in long day-adapted Solanum phureja. Potato Res 41:69–82
Diretto G, Al-Babili S, Tavazza R et al (2007) Metabolic engineering of potato carotenoid content through tuber-specific overexpression of a bacterial mini-pathway. PloS One doi:10.1371/journal.pone.0000350
Dobson G, Griffiths DW, Davies HV, McNicol JW (2004) Comparison of fatty acid and polar lipid contents of tubers from two potato species, Solanum tuberosum and Solanum phureja. J Agric Food Chem 52:6306–6314
Ducreux LJ, Morris WL, Hedley PE et al (2005a) Metabolic engineering of high carotenoid potato tubers containing enhanced levels of beta-carotene and lutein. J Exp Bot 56:81–89
Ducreux LJM, Morris WL, Taylor MA et al (2005b) Agrobacterium-mediated transformation of Solanum phureja. Plant Cell Rep 24:10–14
Ercolano MR, Ballvora A, Paal J et al (2004) Functional complementation analysis in potato via biolistic transformation with BAC large DNA fragments. Mol Breed 13:15–22
Ewing EE, Struik PC (1992) Tuber formation in potato: induction, initiation, and growth. Hort Rev 14:89–198
Faivre-Rampant O, Cardle L, Marshall D et al (2004a) Changes in gene expression during meristem activation processes in Solanum tuberosum with a focus on the regulation of an auxin response factor gene. J Exp Bot 55:613–622
Faivre-Rampant O, Bryan GJ, Roberts AG et al (2004b) Regulated expression of a novel TCP domain transcription factor indicates an involvement in the control of meristem activation processes in Solanum tuberosum. J Exp Bot 55:951–953
Faivre-Rampant O, Gilroy E, Hrubikova K et al (2004c) Potato virus X-induced gene silencing in leaves and tubers of potato. Plant Physiol 134:1308–1316
Fernie AR, Willmitzer L (2001) Molecular and biochemical triggers of potato tuber development. Plant Physiol 127:1459–1465
Gebhardt C (2004) Potato genetics: molecular maps and more. In: Lörz H, Wenzel G (eds) Biotechnology in agriculture and forestry. Springer-Verlag, Berlin, pp 215–224
Geigenberger P, Hajirezaei M, Geiger M et al (1998) Overexpression of pyrophosphatase leads to increased sucrose degradation and starch synthesis, increased activities of enzymes for sucrose–starch interconversions, and increased levels of nucleotides in growing potato tubers. Planta 205:428–437
Ghislain M, Andrade D, Rodriguez F et al (2006) Genetic analysis of the cultivated potato Solanum tuberosum L. Phureja Group using RAPDs and nuclear SSRs. Theor Appl Genet 113:1515–1527
Gilad Y, Oshlack A, Smyth GK, Speed TP, White KP (2006) Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature 440:242–245
Greiner S, Rausch T, Sonnewald U et al (1999) Ectopic expression of a tobacco invertase inhibitor homolog prevents cold-induced sweetening of potato tubers. Nat Biotechnol 17:708–711
Hajirezaei MR, Bornke F, Peisker M et al (2003) Decreased sucrose content triggers starch breakdown and respiration in stored potato tubers (Solanum tuberosum). J Exp Bot 54:477–488
Hannapel DJ (1991) Characterization of the early events of potato tuber development. Physiol Plant 83:568–573
Hofius D, Börnke FAJ (2007) Photosynthesis, carbohydrate metabolism and source-sink relations. In: Vreugdenhil D (ed) Potato biology and biotechnology: advances and perspectives. Elsevier, Amsterdam, The Netherlands, pp 257–285
Hall RD (2006) Plant metabolomics: from holistic hope, to hype, to hot topic. New Phytologist 169:453–468
Jackson SD (1999) Multiple signaling pathways control tuber induction in potato. Plant Physiol 119:1–8
Jackson SD, Heyer A, Dietze J et al (1996) Phytochrome B mediates the photoperiodic control of tuber formation in potato. Plant J 9:159–166
Kalaitzandonakes N, Alston JM, Bradford KJ (2007) Compliance costs for regulatory approval of new biotech crops. Nat Biotechnol 25:509–511
Kamoun S (2007) Groovy times: filamentous pathogen effectors revealed. Curr Opin Plant Biol 10:358–365
Kloosterman B, Vorst O, Hall RD et al (2005) Tuber on a chip: differential gene expression during potato tuber development. Plant Biotech J 3:505–519
Kloosterman B, Navarro C, Bijsterbosch G et al (2007) StGA2ox1 is induced prior to stolon swelling and controls GA levels during potato tuber development. Plant J 52:362–373
Kloosterman B, De Koeyer D, Griffiths R et al (2008) The potato transcriptome: a new look at transcriptional changes during tuber development using the POCI array. Comp Func Genom doi:10.1007/s10142-008-0083-x
Koornneef M, Hanhart CJ, van der Veen JH (1991) A genetic and physiological analysis of late flowering mutants in Arabidopsis thaliana. Mol Gen Genet 229:57–66
Kuang HH, Wei FS, Marano MR et al (2005) The R1 resistance gene cluster contains three groups of independently evolving, type I R1 homologues and shows substantial structural variation among haplotypes of Solanum demissum. Plant J 44:37–51
Lehesranta SJ, Davies HV, Shepherd LVT, Nunan NM et al (2005) Comparison of tuber proteomes of potato (Solanum sp.) varieties, landraces and genetically modified lines. Plant Physiol 138:1690–1699
Lehesranta SJ, Koistinen KM, Massat N et al (2007) Effect of agricultural production systems and their components on protein profiles in potato tubers. Proteomics 7:597–604
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Martinez-Garcia JF, Virgos-Soler A, Prat S (2002) Control of photoperiod-regulated tuberisation in potato by the Arabidopsis flowering-time gene CONSTANS. Proc Natl Acad Sci USA 99:15211–15216
McGregor I (2007) The fresh potato market. In: Vreugdenhil D (ed) Potato biology and biotechnology: advances and perspectives. Elsevier, Amsterdam, pp 3–26
Morris WL, Ducreux L, Griffiths DW et al (2004) Carotenogenesis during tuber development and storage in potato. J Exp Bot 55:975–982
Morris WL, Ducreux LJ, Hedden P et al (2006) Overexpression of a bacterial 1-deoxy-d-xylulose 5-phosphate synthase gene in potato tubers perturbs the isoprenoid metabolic network: implications for the control of the tuber life cycle. J Exp Bot 57:3007–3018
Muth J, Hartje S, Twyman RM et al (2008) Precision breeding for novel starch variants in potato. Plant Biotech J doi:10.1111/j.1467-7652.2008.00340.x
Ooms G, Karp A, Roberts J (1983) From tumour to tuber: tumour cell characteristics and chromosome numbers of crown gall derived tetraploid potato plants (Solanum tuberosum cv Maris Bard). Theor Appl Genet 66:169–172
Paal J, Henselewski H, Muth J (2004) Molecular cloning of the potato Gro1-4 gene conferring resistance to pathotype Ro1 of the root cyst nematode Globodera rostochiensis, based on a candidate gene approach. Plant J 38:285–297
Park T, Gros J, Sikkema A (2005) The late blight resistance locus Rpi-blb3 from Solanum bulbocastanum belongs to a major late blight R gene cluster on chromosome 4 of potato. Mol Plant-Microb Interact 18:722–729
Putterill J, Robson F, Lee K et al (1995) The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 80:847–857
Regan S, Gustafson V, Mallubhotla S et al (2006) Finding the perfect potato: using functional genomics to improve disease resistance and tuber quality traits. Can J Plant Pathol 28:S247–S255
Rodriguez-Falcon M, Bou J, Prat S (2006) Seasonal control of tuberisation in potato: conserved elements with the flowering response. Annu Rev Plant Biol 57:151–180
Römer S, Lubeck J, Kauder F, Steiger S, Adomat C, Sandmann G (2002) Genetic engineering of a zeaxanthin-rich potato by antisense inactivation and co-suppression of carotenoid epoxidation. Metab Eng 4:263–272
Rommens CM, Ye J, Richael C, Swords K (2006) Improving potato storage and processing characteristics through all-native DNA transformation. J Agric Food Chem 54:9882–9887
Rommens CM, Haring MA, Swords K, Davies HV, Belknap WR (2007) The intragenic approach as a new extension to traditional plant breeding. Trends Plant Sci 12:397–403
Scholz M, Gatzek S, Sterling A, Fiehn O, Selbig J (2004) Metabolite fingerprinting: detecting biological features by independent component analysis. Bioinformatics 20:2447–2454
Schouten HJ, Krens FA, Jacobsen E (2006) Cisgenic plants are similar to traditionally bred plants: international regulations for genetically modified organisms should be altered to exempt cisgenesis. EMBO Rep 7:750–753
Shepherd T, Dobson G, Marshall R et al (2007) Profiling of metabolites and volatile flavour compounds from Solanum species using gas chromatograph-mass spectrometry. In: Vreugdenhil D (ed) Potato biology and biotechnology: advances and perspectives. Elsevier, Amsterdam, pp 209–219
Sonnewald U (2001) Control of potato tuber sprouting. Trends Plant Sci 6:333–335
Spooner DM, Nunez J, Trujillo G et al (2007) Extensive simple sequence repeat genotyping of potato landraces supports a major reevaluation of their gene pool structure and classification. Proc Natl Acad Sci USA 104:19398–19403
Struik PC, Haverkort AJ, Vreugdenhil D et al (1990) Manipulation of tuber-size distribution of a potato crop. Potato Res 33:417–432
Struik PC, Vreugdenhil D, Haverkort AJ et al (1991) Possible mechanisms of hierarchy among tubers from one stem of a potato (Solanum tuberosum L.) plant. Potato Res 34:187–203
Suttle JC (2004) Physiological regulation of potato tuber dormancy. Am J Potato Res 81:253–262
Urbanczyk-Wochniak E, Luedemann A, Kopka AR et al (2003) Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Rep 4(10):989–993
Van den Berg JH, Ewing EE, Plaisted RL et al (1996) QTL analysis of potato tuberisation. Theor Appl Genet 93:307–316
Van der Hoeven R, Ronning C, Giovannoni J et al (2002) Deductions about the number, organization, and evolution of genes in the tomato genome based on analysis of a large expressed sequence tag collection and selective genomic sequencing. Plant Cell 14:1441–1456
Van der Vossen EAG, van der Voort JNAMR, Kanyuka K et al (2000) Homologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: a virus and a nematode. Plant J 23:567–576
Viola R, Roberts AG, Haupt S et al (2001a) Tuberisation in potato involves a switch from apoplastic to symplastic phloem unloading. Plant Cell 13:385–398
Viola R, Taylor M, Oparka K (2001b) Meristem activation in potato: impact on tuber formation, development and dormancy. Annual Report of the Scottish Crop Research Institute 2000/2001, SCRI, Dundee, pp 99–102
Vreugdenhil D, Struik PC (1989) An integrated view of the hormonal regulation of tuber formation in potato (Solanum tuberosum L.). Physiol Plant 75:525–531
Weigel D, Ahn JH, Blazquez MA et al (2000) Activation tagging in Arabidopsis. Plant Physiol 122:1003–1013
Zhang L, Hausler RE, Greiten C et al (2008) Overriding the co-limiting import of carbon and energy into tuber amyloplasts increases the starch content and yield of transgenic potato plants. Plant Biotech J 6:453–464
Acknowledgement
The authors wish to thank the Scottish Government for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davies, H., Bryan, G.J. & Taylor, M. Advances in Functional Genomics and Genetic Modification of Potato. Potato Res. 51, 283–299 (2008). https://doi.org/10.1007/s11540-008-9112-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11540-008-9112-3