
Abstract
Respiratory viral infections are common in the general population and one of the most important causes of asthma aggravation and exacerbation. Despite many studies, it is not well understood how viral infections cause more severe symptoms and exacerbations in asthmatics. We develop a mathematical model of two types of macrophages that play complementary roles in fighting viral infection: classically \((\hbox {CA}\)-\(\hbox {M}\Phi )\) and alternatively activated macrophages \((\hbox {AA}\)-\(\hbox {M}\Phi )\). \(\hbox {CA}\)-\(\hbox {M}\Phi \) destroy infected cells and tissues to remove viruses, while \(\hbox {AA}\)-\(\hbox {M}\Phi \) repair damaged tissues. We show that a higher viral load or longer duration of infection provokes a stronger immune response from the macrophage system. By adjusting the parameters, we model the differences in response to respiratory viral infection in normal and asthmatic subjects and show how this skews the system toward a response that generates more severe symptoms in asthmatic patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Viral respiratory infections are among the most common human diseases and can be caused by influenza virus, parainfluenza virus, adenovirus, respiratory syncytial virus (RSV), human rhino virus (HRV) among others (Syrmis et al. 2004; Weigl et al. 2000). Collectively, these viruses account for more than 80% of identified lower respiratory infections in children (Erdman et al. 2003) and are responsible for more serious complications in children, the elderly, and immunocompromised patients (Syrmis et al. 2004). Respiratory viral infections also aggravate symptoms and trigger exacerbations in patients with asthma or chronic obstructive pulmonary disease (COPD) (Corne et al. 2002; Jackson and Johnston 2010; Proud and Chow 2006).
Most respiratory viruses infect airway epithelial cells (Proud and Chow 2006). Immediately after infection, viruses are recognized and activate the cells via Toll-like receptors (TLR) (Boukhvalova et al. 2006). Internalized viruses also activate intracellular signaling systems through the NF-kB (nuclear factor-kB) pathway, resulting in production of type I interferons, such as \(\hbox {IFN}\)-\(\alpha \) and \(\hbox {IFN}\)-\(\beta \), that inhibit replication and spreading of viruses (Takaoka et al. 2003).
After epithelial cells have been infected and begun to respond, alveolar macrophages take control of the defense system. Monocyte-derived macrophages can then differentiate into M1 and M2 macrophages (Hussell and Bell 2014; Kim et al. 2013; Shirey et al. 2010). Differentiation into M1 macrophages, often called classically activated macrophages \((\hbox {CA}\)-\(\hbox {M}\Phi )\), requires both pro-inflammatory stimuli, such as \(\hbox {IFN}\)-\(\gamma \), and activation by pathogen-associated molecules (Hussell and Bell 2014). \(\hbox {CA}\)-\(\hbox {M}\Phi \) differentiation can also be induced by stimulation with IL-12 (Hussell and Bell 2014; Kim et al. 2013; Shirey et al. 2010). \(\hbox {CA}\)-\(\hbox {M}\Phi \) produce pro-inflammatory molecules such as \(\hbox {TNF}\)-\(\alpha \), iNOS, and COX-2 and initiate inflammation designed to remove the antigens, but which can also damage host tissue (Rath et al. 2014; Richardson et al. 2005). The enzyme iNOS generates nitric oxide (NO), which can be detected in the exhaled air from normal human subjects with upper respiratory tract infections (Kharitonov et al. 1995). Exhaled NO is associated with chronic inflammation in asthmatic airways and exacerbated asthma. In addition to fighting infections, NO can damage infected cells and structures. For example, inflammation and tissue injury in the lungs of mice infected by Sendai virus were significantly lower in iNOS-deficient than in wild-type mice (Akaike et al. 2000). Less severe pneumonitis was observed in \(\hbox {NOS2}^{-/-}\) mice than in \(\hbox {NOS2}^{+/+}\) mice (Karupiah et al. 1998).
M2 macrophages, often called alternatively activated macrophages \((\hbox {AA}\)-\(\hbox {M}\Phi )\), are associated with tissue repair. \(\hbox {AA}\)-\(\hbox {M}\Phi \) differentiate in response to stimulus by IL-4, IL-13 (Shaykhiev et al. 2009), or \(\hbox {IFN}\)-\(\beta \) (Shirey et al. 2010), and enhanced by activation and up-regulation of IL-4 receptor-\(\alpha \) (\(\hbox {IL}\)-\(4\hbox {R}\alpha )\). \(\hbox {AA}\)-\(\hbox {M}\Phi \) encourage repair of damaged tissue by producing arginase-1 (Hussell and Bell 2014; Shirey et al. 2010). Arginase-1 competes with iNOS for arginine to produce L-ornithine and urea, rather than NO (Rath et al. 2014). Ornithine is metabolized to proline, a substrate for collagen synthesis, and polyamines, that stimulate cellular proliferation. This pathway is important in the healing process (Curran et al. 2006), and thus, \(\hbox {AA}\)-\(\hbox {M}\Phi \) potentially protect cells from the structural damage associated with viral invasion and control. Conversely, iNOS generated by \(\hbox {CA}\)-\(\hbox {M}\Phi \) induce the production of redox molecules in addition to NO, such as NOHA (N-Hydroxyarginine), that inhibit arginase (Wink et al. 2011).
Asthma is characterized by respiratory symptoms and reversible airway obstruction caused by chronic airway inflammation (GINA 2014). There have been relatively few mathematical models of asthma. Due to the complexity of the disease, those models have addressed very different questions with contrasting methods. A agent-based model of the interactions between innate and adaptive immunity that reproduces hypersensitivity to RSV and HRV infections was introduced (Folcik et al. 2007) and extended by adding Th17 cells and recruitment of dendritic cells (Song et al. 2012). A model of airway smooth muscle in asthma showed that consistent inflammatory signals promote the proliferation that is linked to asthma progression (Chernyavsky et al. 2014). Kim et al. focused on the Th1/Th2 balance that responds to LPS exposure (Kim et al. 2013). This model includes only a single type of macrophage that is suppressed by IL-4 and thus corresponds to the classically activated macrophages in our model. Our modeling approach provides a complement by looking at a switch upstream from the Th1/Th2 system to capture the macrophage dynamics soon after infection which control inflammation, damage, and the adaptive immune response.
Respiratory viral infections cause a variety of clinical syndromes from the upper respiratory tract infection with rhinitis to the lower respiratory tract disease with bronchiolitis and pneumonia. If airway inflammation progresses sufficiently, wheezing in children may occur, which is associated with later asthma (Jackson et al. 2008). The severity of symptoms depends on the level of inflammation of airway (Dolin 2015). Failure to induce \(\hbox {AA}\)-\(\hbox {M}\Phi \) prolongs expression of the \(\hbox {CA}\)-\(\hbox {M}\Phi \) phenotype and generates more peribronchiolar and perivascular inflammation in an animal model (Shirey et al. 2010). Clinical symptom severity is associated with the initial virus load and the time needed to clear the virus (DeVincenzo et al. 2010; Saleeby et al. 2011).
Respiratory viral infections are a common cause of asthma exacerbations showing a strong positive relationship between epidemic seasonal upper respiratory tract infection and increases in hospitalizations for exacerbations of asthma (Proud and Chow 2006). Asthmatics have more severe and long-lasting lower respiratory tract symptoms after viral respiratory infections (Busse et al. 2010; Corne et al. 2002). Viral respiratory infections can lead to increased airway responsiveness in subjects with asthma, possibly through effects on neural control of airway function (Jacoby 2004).
Despite these studies, the mechanisms by which respiratory viruses trigger asthma exacerbations remain incompletely understood, including complex interactions among host, viral, and environmental factors. In the context of this model, differences in production of \(\hbox {IFN}\)-\(\beta \) and IL-4 in asthmatics after viral infection provide two testable mechanisms. Primary human bronchial epithelial cells from asthmatics show lower production of virus-induced \(\hbox {IFN}\)-\(\beta \) (Dougherty and Fahy 2009; Wark et al. 2009, 2005). In addition, the level of IL-4 is increased in BAL fluid from atopic asthmatics after allergen challenge (Virchow et al. 1995). These findings are not universal, with one study finding IL-4 to be difficult to detect in asthmatics regardless of their symptoms (Broide et al. 1992), and another finding IL-4 and \(\hbox {IFN}\)-\(\gamma \) levels in BAL fluid to be similar in patients with asthma, bronchiolitis induced by RSV, and normal control (Kim et al. 2003).
From this biological and clinical background, we derive three hypotheses about how viral infections alter the balance of the alveolar macrophage system and potentially trigger asthma exacerbations:
-
1.
A higher viral load will elicit a stronger immune response,
-
2.
Longer duration of infection will elicit a stronger immune response,
-
3.
A dysregulated immune response to viral infection will be elicited in asthma-like conditions.
In this paper, we develop a mathematical model of the interaction between respiratory viruses and the immune system to predict the activation of two types of macrophages to test these hypotheses. We focus on how respiratory viral infection changes the balance between \(\hbox {CA}\)-\(\hbox {M}\Phi \) and \(\hbox {AA}\)-\(\hbox {M}\Phi \), how this response differs in hosts with asthma-like conditions, and how those differences can lead to accentuated symptoms.
2 Methods
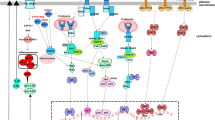
We use ordinary differential equations to track populations of viruses and cells, and the concentrations of key cytokines and enzymes secreted by the cells. Our model considers three cell types: airway epithelial cells, classical activated macrophages \((\hbox {CA}\)-\(\hbox {M}\Phi )\), and alternatively activated macrophages \((\hbox {AA}\)-\(\hbox {M}\Phi )\); two cytokines: \(\hbox {IFN}\)-\(\beta \) and IL-4; and two enzymes: iNOS and arginase-1 (Fig. 1; Table 1). Numerical solutions of the model were performed with deSolve package in R (R Development Core Team 2008; Soetaert et al. 2010). We describe the model in three steps, first the viral dynamics, then the dynamics of epithelial cells and the interferon they produce, and finally the macrophages and their products.

State variables, transitions, and productions in the model. The names of variables and parameters are given in Table 1. Solid arrows indicate transitions of cells, dashed arrows indicate activations or productions, and dashed lines with vertical ends indicate inhibitions. V Virus, Ed dormant airway epithelial cell, Ea activated airway epithelial cell, Md dormant macrophage, Mc \(\hbox {CA}\)-\(\hbox {M}\Phi \) or classically activated macrophages, Ma \(\hbox {AA}\)-\(\hbox {M}\Phi \) or alternatively activated macrophages, \({{ IFN}_b}\) \(\hbox {INF}\)-\(\beta \), IL4 IL-4, iNOS inducible nitric oxide synthase, Arg arginase-1
Viral Dynamics After infection with inoculum \(V_{\mathrm{min}}\) at time \(t_{\mathrm{start}}\), we assume that the virus population increases exponentially to a peak of \(V_{\mathrm{max}}\) at time \(t_{\mathrm{peak}}\) and then declines exponentially. Because we do not include the full complexity of the immune response, we model the virus as following the two-phase exponential solution that accurately approximates more detailed models of virus dynamics (Smith et al. 2010). We parameterize the virus dynamics in terms of the maximum load of virus (\(V_{\mathrm{max}}\)) and timing of the infection.
Here, \({\beta }_\mathrm{V}\) is the intrinsic replication rate of the virus and \({\delta }_\mathrm{V}\) its loss rate that could depend on the immune response. The value of \({\beta }_\mathrm{V}\) is chosen to ensure that \(V(t_{\mathrm{start}} +t_{\mathrm{peak}} )=V_{\mathrm{max}}\). The duration of infection is defined as \(t_{\mathrm{peak}} +\frac{1}{{\delta }_\mathrm{V}}\).
Airway Epithelial Cells and IFN-\(\beta \) The next set of equations describe the airway epithelial cells directly infected by the virus and the type I interferon they produce. In our model, these cells can be in two states, dormant (Ed) and activated (Ea). Ed transition to the activated state upon exposure to virus at rate \(\alpha _{\mathrm{E}}\), while Ea return to the dormant state at rate \(\gamma \). Ed turn over at rate \(\delta _{\mathrm{Ed}}\) and remain at the equilibrium \(\hbox {Ed}_{\mathrm{0}}\) in the absence of virus. Ea die at the accelerated rate of \(\delta _{\mathrm{Ea}}\) and produce \(\hbox {IFN}\)-\(\beta \,\,(\hbox {IFN}_{\mathrm{b}})\) at rate \(c_{\mathrm{b}}\). In the absence of viral infection, the level of \(\hbox {IFN}_{\mathrm{b}}\) remains at the equilibrium \(\hbox {IFN}_{\mathrm{b0}}\) with turnover rate \(\delta _{\mathrm{IFN_b}}\).
Classically and Alternatively Activated Macrophages and IL-4
The balance between classically activated macrophages (Mc) and alternatively activated macrophages (Ma), as controlled by the cytokines \(\hbox {IFN}_{\mathrm{b}}\) and IL4, is at the heart of our model. The key switch occurs after dormant macrophages are activated at rate \({\alpha }_{\mathrm{M}}\) by exposure to virus. We let \(f_{a}\) be the fraction of macrophages that become \(\hbox {AA}\)-\(\hbox {M}\Phi \) upon activation. RSV failed to induce expression of \(\hbox {AA}\)-\(\hbox {M}\phi \) genes in \(\hbox {IL}\)-\(\hbox {4R}\alpha ^{-/-}\) macrophages and partially failed in \(\hbox {IFN}\)-\(\beta ^{-/-}\) macrophages (Shirey et al. 2010). Because the differentiation of \(\hbox {AA}\)-\(\hbox {M}\phi \) depends on \(\hbox {IFN}\)-\(\beta \) and IL-4, we set this fraction to depend on their product according to
We make several additional assumptions related to differentiation into \(\hbox {CA}\)-\(\hbox {M}\Phi \) and \(\hbox {AA}\)-\(\hbox {M}\Phi \). To model response to viral infection, we do not track macrophages that have previously polarized to \(\hbox {CA}\)-\(\hbox {M}\Phi \) or \(\hbox {AA}\)-\(\hbox {M}\Phi \) and assume that macrophages differentiate only to one of \(\hbox {CA}\)-\(\hbox {M}\Phi \) and \(\hbox {AA}\)-\(\hbox {M}\Phi \) so that the fraction of \(\hbox {CA}\)-\(\hbox {M}\Phi \) is \(1-f_{\mathrm{a}}\) (Eq. 5). This is a simplification; \(\hbox {CA}\)-\(\hbox {M}\Phi \) appear earlier than \(\hbox {AA}\)-\(\hbox {M}\Phi \) (Shirey et al. 2010) because their differentiation needs only viral or bacterial stimulation, while \(\hbox {AA}\)-\(\hbox {M}\Phi \) need additional stimulation by the cytokines \(\hbox {IFN}\)-\(\beta \) and IL-4. Our model does not include this time delay.
In the absence of infection, dormant macrophages, Md, remain at the equilibrium \(\hbox {Md}_\mathrm{0}\) with turnover rate \({\delta }_{{\mathrm{Md}}} \). Mc and Ma die at rates \(\delta _{\mathrm{Mc}}\) and \(\delta _{\mathrm{Ma}}\), respectively. Estimates of the life span of alveolar macrophages in normal conditions range from 2 weeks (van oud Alblas and van Furth 1979) to 8 months (Murphy et al. 2008). The life span of activated \(\hbox {CA}\)-\(\hbox {M}\Phi \) and \(\hbox {AA}\)-\(\hbox {M}\Phi \) is not known.
We track three of their chemical products: IL4, iNOS, and arginase-1. \(\hbox {AA}\)-\(\hbox {M}\Phi \) produce IL4 at rate \(c_{4}\) and IL4 remains at the equilibrium \(\hbox {IL4}_{\mathrm{0}}\) with turnover rate \({\delta }_{{\mathrm{IL}}4}\). Due to their mechanisms of inhibition, the equations for iNOS (NOS) and arginase-1 (Arg) are more complex. iNOS is produced both by \(\hbox {CA}\)-\(\hbox {M}\Phi \) and by activated epithelial cells (Tsutsumi et al. 1999), with rates \(C_{{\mathrm{NOSm}}} \hbox { and } C_{{\mathrm{NOSe}}}\), respectively, although these relative values are unknown. Arginase-1 is produced by \(\hbox {AA}\)-\(\hbox {M}\Phi \) at rate \(C_{{\mathrm{Arg}}} \). To capture the mechanisms of inhibition between NOS and Arg, we decrease NOS production by the factor of \(1+\Phi \cdot \hbox {Ma}\), where \(\Phi \) represents the IL-10 produced by \(\hbox {AA}\)-\(\hbox {M}\Phi \) which suppresses iNOS gene transcription (Modolell et al. 1995). NOHA (N-Hydroxyarginine), one of the products induced by NOS, inhibits arginase (Wink et al. 2011). We do not explicitly include NOHA, but reduce Arg production by a factor \(1+\Phi _\mathrm{A}\cdot \hbox {NOS}\).
Parameter Estimates Because direct estimates of most of the parameters are not available, we use basic properties of the virus and the immune system to develop order of magnitude estimates as a starting point. The number of alveolar epithelial cells and macrophages is variable in species and experiments. Our initial conditions for dormant epithelial cells (Ed) and dormant macrophage (Md) come from a rough estimate of one Md per 1000 Ed. Results are not sensitive to this choice because it is an effective number than can be absorbed into the rates of production of IL4, NOS and Arg. The normal level of \(\hbox {IFN}\)-\(\beta \) in the alveolus in normal healthy subjects is not known. Little or no \(\hbox {IFN}\)-\(\beta \) is found in the serum even after administration of synthetic \(\hbox {IFN}\)-\(\beta \) to healthy volunteers with a clinically recommended dose (Sturzebecher et al. 1999). The exact half-life of \(\hbox {IFN}\)-\(\beta \) is also uncertain, with a reported serum half-life of \(\hbox {IFN}\)-\(\beta \) ranging from minutes to hours (McKenna et al. 2004). The level of IL-4 has been known to change according to subject’s age (Kleiner et al. 2013), and appropriate ratio of IL-4 to \(\hbox {IFN}\)-\(\beta \) remains unknown, as do normal levels of arginase-1 and iNOS. The half-life of iNOS is not known in macrophages, but is \(1.6 \pm 0.3\) h in primary bronchial epithelial cells (Kolodziejski et al. 2004).
Given this uncertainty, we calibrate the unknown parameters based on results of in vivo experiments (Shirey et al. 2010). This experiment observed \(\hbox {CA}\)-\(\hbox {M}\Phi \) and \(\hbox {AA}\)-\(\hbox {M}\Phi \) after RSV infection in alveolar or peritoneal macrophages and measured the products (including IL-4, iNOS, COX-2, and arginase-1) that represent each type. We used the observed relative changes of IL-4, arginase-1, and iNOS from alveolar macrophages and of \(\hbox {INF}\)-\(\beta \) from peritoneal macrophages as our target values (Fig. 1a, b and suppl. Fig. S6-A in Shirey et al. 2010; Table 2). The target values are the factor by which that value increased relative to the initial conditions at time points 25, 50, and 75 h after inoculation with virus.
To assess which model predictions could be effectively constrained by the available data, we identified an ensemble of parameter combinations consistent with the data. As our measure of deviation, we used the sum of the squared differences between the log-transformed simulation values with the targets found from the literature (the fold increases in Table 2). Starting with estimates of 30 initial conditions and parameters, we tested 7 values of one randomly chosen parameter ranging from a factor of 10 below to a factor of 10 above the current value, choosing the next value that minimized the difference from the target. We repeated this procedure 25 times, choosing the next parameter, with replacement, to test with equal probability. After these 25 tests, we reduced the range of the search by a factor of 1.2. We conducted 30 sets of 25 tests, after which the total deviation stopped decreasing. To generate each set of parameters, we thus ran a total of 5250 simulations. The initial conditions for Ea, Mc and Ma were kept at 0 throughout. We did not vary the value of \(\delta _{\mathrm{V}}\) because we assumed a fixed duration of viral infection. We followed this procedure to obtain 50 parameter sets that converged to a deviation of less than 0.02. This corresponds to a deviation of 1% from each of the 12 targets in Table 2 and guarantees that the simulation differs from no target by more than a factor of 1.2. The best-fitting set of parameters that created the minimum deviation of 0.012 was chosen for the baseline or normal condition in the next step of analyze.
Sensitivity Analysis We present sensitivity analysis in two ways. First, we illustrate the dynamics resulting from 50 sets of parameter values that converged to closely match the target values to show which state variables can and cannot be resolved (Fig. 2). Second, we show the distribution of each parameter in this set to show which parameters are best resolved (Fig. 3).

Dynamics with 50 sets of parameter values that closely match the target values in Table 2, with variable names as in Table 1 and Fig. 1. Darker colors indicate closer fits. The horizontal axis represents time in hours, vertical axis counts and concentrations as in Table 1. Dashed vertical lines indicate the start, peak, and end of infection

The log 10 transformed distributions of parameter values in our set of 50 that closely match the target values, ordered by increasing range. Names of variables and parameters are as in Table 1. Parameters are classified as follows: Epi (epithelial cells) \(\hbox {Ed}_{\mathrm{0}}\), \(\delta _{\mathrm{Ed}}\), \(\delta _{\mathrm{Ea}}\), \(\alpha _{\mathrm{E}}\), \(\gamma \); Mac (macrophages) \(\hbox {Md}_{\mathrm{0}}\), \(\alpha _{\mathrm{M}}\), \(\delta _{\mathrm{Md}}\), \(\delta _{\mathrm{Mc}}\), \(\delta _{\mathrm{Ma}}\); \({IFN}_b\) \(\hbox {IFN}_\mathrm{b0}\), \(c_{\mathrm{b}}\), \(\uplambda _{\mathrm{4}}\), \(\delta _{\mathrm{IFN_b}}\); IL4 \(\hbox {IL4}_{\mathrm{0}}\), \(c_{\mathrm{4}}\), \(\delta _{\mathrm{IL4}}\); NOS \(\hbox {NOS}_{\mathrm{0}}\), \(c_{\mathrm{NOSm}}\), \(c_{\mathrm{NOSe}}\), \(\delta _{\mathrm{NOS}}\), \(\Phi \); Arg \(\hbox {Arg}_{\mathrm{0}}\), cArg, \(\delta _{\mathrm{Arg}}\), \(\Phi _{\mathrm{A}}\); Virus \(V_{\mathrm{min}}\), \(V_{\mathrm{max}}\), \(t_{\mathrm{peak}}\), \({\varvec{\varDelta }} t(t_{\mathrm{peak}} - t_{\mathrm{start}})\) (Color figure online)
Asthma-Like Conditions Primary human bronchial epithelial cells (HBEC) from asthmatics show impairment of virus-induced \(\hbox {IFN}\)-\(\beta \) mRNA expression, with production of \(\hbox {IFN}\)-\(\beta \) decreased to approximately 40% of normal (Wark et al. 2009, 2005). Type I IFN was also decreased in PBMCs from children (Iikura et al. 2011), and in HBECs and bronchoalveolar lavage (BAL) cells (Khaitov et al. 2009). As a consequence, arginase-1 did not increase in \(\hbox {IFN}\)-\(\beta \) KO mice as much as in wild-type mice, indicating decreased differentiation of \(\hbox {AA}\)-\(\hbox {M}\Phi \) (Shirey et al. 2010).
The percentage of IL-4 mRNA+ cells in BAL fluid or bronchial tissue is significantly larger in asthmatics than in normal controls, with the mean value for IL-4 increased roughly 1.5 times more in BAL fluid after allergen challenge (Virchow et al. 1995). The normal condition is composed of best-fitting parameter set. We model these conditions by adjusting the \(\hbox {IFN}\)-\(\beta \) production rate \((c_\mathrm{b})\) and the IL-4 production rate \((c_\mathrm{4})\). We tested two conditions: reducing the \(\hbox {IFN}\)-\(\beta \) production rate \((c_\mathrm{b})\) by 50% and doubling the IL-4 production rate \((c_\mathrm{4})\).
3 Results
3.1 Parameter Estimation and Sensitivity Analysis
The parameter values that provide the closest fit to the targets are given in Table 1. The ability of this model to resolve the dynamics is shown in Fig. 2, and the resolution of each parameter value and initial condition is shown in Fig. 3. Our trajectories show that the dynamics are well resolved for all state variables except Mc and NOS, with our calibration targets insufficient to predict whether these will decline rapidly, slowly, or not at all. Most parameters are reasonably well resolved, except for clearance of Mc and Ed (Mc and Ed), the half-saturation constant in the equation for the fraction of macrophage that differentiate to Ma (\(f_{a}\)), and the production rate of NOS by Mc (\(c_\mathrm{NOSm}\)).
3.2 The Response of the Macrophage System to Viral Stimulation
All activated cells and their products increase at the start of infection (Fig. 2). Ea and \(\hbox {IFN}_\mathrm{b}\) peak quickly and decline to baseline. Because \(\hbox {M}\Phi \) activate more slowly, they produce first a burst of Mc, then Ma, and then their products IL4 and Arg. Ea, \(\hbox {IFN}_\mathrm{b}\) and IL4 largely resolve before the end of viral infection, while Mc and NOS persist well beyond.
3.3 The Effects of Viral Load and Duration of Infection
To test the effects of viral load, we solve the model with a \(V_{\mathrm{max}}\) value of double or half of the baseline value (Fig. 4). Higher viral loads induce slightly greater increases in the populations of all inflammatory cells and their products, but without changing the duration of the \(\hbox {IFN}_\mathrm{b}\) and IL-4 response. We do predict a long-term increase in Mc and NOS, but this occurs in the most poorly resolved portion of the prediction. To test the effect of duration of infection, we solve the model with values of \(t_\mathrm{peak}\) double and half that of our best estimate of 40.6 h (Fig. 5). We predict that a longer duration infection delays most responses only slightly and produces greatly reduced peaks only in Ea and \(\mathrm{IFN_b}\), an initially lower but eventually higher Mc and NOS, and slightly higher levels of IL-4 and Arg.

Effects of viral load. Notation and axes are as in Fig. 2 with time in hours but with \(V_{\mathrm{max}}\) increased to double (red curve) or half (blue curve) of its baseline value (black curve). The baseline represents the condition that uses the best-fitting parameter values as in Table 1. Dashed vertical lines indicate the start, peak, and end of infection (Color figure online)

Effects of duration of infection load. Notation and axes are as in Fig. 2 with time in hours but with \({V}_{\mathrm{max}}\) increased to double (red curve) or half (blue curve) of its baseline value (black curve). The baseline represents the condition that uses the best-fitting parameter values as in Table 1. The dashed vertical lines indicate the start of infection and the solid vertical lines the peak of infection with the three values of \(t_{\mathrm{peak}}\) (Color figure online)
4 The Effects of Asthma-Like Conditions
In addition to lower \(\hbox {IFN}_{\mathrm{b}}\), decreased \(\hbox {IFN}_{\mathrm{b}}\) production \((c_{\mathrm{b}})\) leads primarily to increased Mc and NOS production (Fig. 6). Increased IL4 production \((c_{4})\) produces of course higher IL4 and then lower Mc and NOS production with little change in the other outputs (Fig. 7). Only the first of these conditions is associated with a higher ratio of NOS to Arg and thus with the most damaging inflammation.

Effects of asthma-like condition modeled by adjusting the \(\hbox {IFN}\)-\(\beta \) production rate \((c_\mathrm{b})\). Dashed vertical lines indicate the start, peak, and end of infection, with all times in hours. The baseline (black curve) represents the normal condition that uses the best-fitting parameter values as in Table 1. Fifty percent reduction in the \(\hbox {IFN}\)-\(\beta \) production rate, modeling an asthma-like condition (blue curve), leads primarily to increased Mc and NOS production relative to the baseline or doubled production (red curve) (Color figure online)

Effects of asthma-like condition modeled by adjusting the IL-4 production rate \((c_{4})\). Dashed vertical lines indicate the start, peak, and end of infection with all times in hours. The baseline (black curve) represents the normal condition with the best-fitting parameter values from Table 1. Increased IL-4 production rate (red curve) models the asthma-like condition that produces higher IL4 in the short run and lower Mc and NOS production over the longer run relative to the baseline or decreased production (blue curve) (Color figure online)
5 Discussion
We develop a mathematical model of the macrophage response to respiratory viral infection and use it to perform virtual experiments by manipulating the strength and duration of viral infection, and patient parameters that predispose to asthma. The macrophage system in the model consists of classically activated \((\hbox {CA}\)-\(\hbox {M}\Phi )\) and alternatively activated macrophages \((\hbox {AA}\)-\(\hbox {M}\Phi )\), and their respective products, iNOS and arginase-1. The balance between these two types of macrophage plays an important role in tissue destruction or repair, because \(\hbox {CA}\)-\(\hbox {M}\Phi \) are associated with viral clearance and tissue damage, while \(\hbox {AA}\)-\(\hbox {M}\Phi \) are associated with tissue repair. We model this balance as being controlled by \(\hbox {IFN}\)-\(\beta \) produced by epithelial cells and IL-4 produced by \(\hbox {AA}\)-\(\hbox {M}\Phi \).
Despite only limited available data to calibrate, we found that sets of parameters that fit these data resolve the dynamics for all of the state variables well except for \(\hbox {CA}\)-\(\hbox {M}\Phi \) and NOS, which are consistently predicted to increase quickly during the infection but may or may not decrease or rebound thereafter (Fig. 2). Longer-term data on responses to infection are needed to determine this component of the dynamics. The most poorly resolved parameters are those for the death of \(\hbox {CA}\)-\(\hbox {M}\Phi \) and of dormant epithelial cells (Fig. 3). In the available data (Shirey et al. 2010), \(\hbox {CA}\)-\(\hbox {M}\Phi \) can be only distinguished by the expression of their markers, in this case NOS for \(\hbox {CA}\)-\(\hbox {M}\Phi \) and arginase for \(\hbox {AA}\)-\(\hbox {M}\Phi \). These targets were measured within 72 h of infection, but we solved the model well beyond this time, leading in part to the regions of poor model resolution.
We found that higher viral load or longer duration of infection period primarily increases the level of \(\hbox {CA}\)-\(\hbox {M}\Phi \) with a smaller effect on \(\hbox {AA}\)-\(\hbox {M}\Phi \). An increased magnitude and duration of \(\hbox {CA}\)-\(\hbox {M}\Phi \) are thought to be caused more tissue destruction, and the model predicts more severe and longer duration symptoms, but requires more data to predict the long-term progression. Mechanistically, the increase of \(\hbox {CA}\)-\(\hbox {M}\Phi \) generates more iNOS, which induces higher production of cytotoxic nitric oxide, NO (Wink et al. 2011). The higher numbers of infected epithelial cells produce a strong short-term \(\hbox {IFN}\)-\(\beta \) and NO response, while the accentuated \(\hbox {AA}\)-\(\hbox {M}\Phi \) response predicts more arginase-1 and potentially asthma-inducing IL-4.
Based on experimental measurements, we modeled asthma-like conditions as a decrease in virus-induced production of \(\hbox {IFN}\)-\(\beta \) (Dougherty and Fahy 2009; Iikura et al. 2011; Wark et al. 2009, 2005) and an increase in IL-4 production (Broide et al. 1992; Hussell et al. 1996; Kim et al. 2003). Deficiency in interferon is likely related to higher levels of viral replication in asthmatics (Contoli et al. 2006; Gehlhar et al. 2006). We present two virtual conditions of asthma-like conditions: decreased production of \(\hbox {IFN}\)-\(\beta \) (Fig. 6) and increased production of IL-4 (Fig. 7). The key result is the opposite responses of \(\hbox {CA}\)-\(\hbox {M}\Phi \) compared to normal responses. Reduced production of \(\hbox {IFN}\)-\(\beta \) predicts a greater total population of \(\hbox {CA}\)-\(\hbox {M}\Phi \) and a smaller population of \(\hbox {AA}\)-\(\hbox {M}\Phi \), leading to higher iNOS (Fig. 6). This in turn implies more destruction and less repair, and potentially more severe symptoms and even exacerbations in asthma patients with a respiratory viral infection. Our model predicts reduced Mc and NOS with a high value of IL-4 production (Fig. 7).
Because this model captures only a few components of the full complexity of asthma, we describe our experiments as “asthma-like conditions.” Asthma is diagnosed by a history of respiratory symptoms, such as cough, shortness of breath, and wheeze, that vary over time and in intensity, and the evidence of variable expiratory airflow limitation documented from bronchodilator reversibility test (GINA 2014). In addition, asthma is a heterogeneous disease that divides into different clinical phenotypes based on the age of onset, presence of obesity, decreased lung function, presence of atopy or eosinophilia, and particular biomarkers (Haldar et al. 2008; Moore et al. 2009). This model captures only one aspect of these many pathogenic syndromes.
Our model breaks macrophage into two discrete types, but recent work suggests a continuum of phenotypes, with macrophages are able to switch from the repair pathway to the inflammation pathway (but not in reverse), a transition missing from our model (Childs et al. 2011). For example, a switch from \(\hbox {AA}\)-\(\hbox {M}\Phi \) to \(\hbox {CA}\)-\(\hbox {M}\Phi \) activation in the inflammatory lesion was observed in an atherosclerosis model, although the possibility of a change in the cytokine milieu was not considered in this study (Khallou-Laschet et al. 2010).
This model makes many other simplifying assumptions. Like most virus dynamics models, we do not consider space and migration of cells. Effectively, our model tracks the virus-induced responses in one alveolus. For simplicity, we do not distinguish IL-4 and IL-13. These two Th2 cytokines have structural similarity, share the receptor subunit \(\hbox {IL}\)-\(\hbox {4R}\alpha \), and activate common signaling pathways (Jiang et al. 2000). The primary sources of IL-4 are basophils, eosinophils, NK cells, and Th2 lymphocytes (Kim et al. 2013), and our model assumes that these cell populations are proportional to the \(\hbox {AA}\)-\(\hbox {M}\Phi \) population and thus can be incorporated the production into the parameter \(c_{4}\). Similarly, macrophages are not the only cells to produce NO in the upper and lower respiratory tract, and exhaled NO is more diagnostic of eosinophilic inflammation in the airway than inflammation induced by macrophages (Dweik et al. 2011). Elevated NO concentration not only helps with clearance of virus but also inhibits normal monocyte growth and platelet aggregation (Wink et al. 2011). \(\hbox {CA}\)-\(\hbox {M}\Phi \) also produce COX-2 and other pro-inflammatory cytokines, such as IL-6, \(\hbox {TNF}\)-\(\alpha \), and IL-1, which can create widespread ramifications like tissue destruction and increased symptoms with their many unwanted clinical consequences during clearance of virus (Shirey et al. 2010; Wink et al. 2011). Normal levels of most of cytokines and enzymes, including target products, are not known.
We used fold change from initial value after viral stimulation to obtain estimates of all parameter values. Therefore, values of all cells and products do not reflect the absolute measured values in a real situation. The purpose of this study is not to predict the exact values of cytokine or cell levels, but the changing balance between two types of macrophage after viral infection in normal and asthma-like conditions.
Our model bases the course of viral loads on humans infected experimentally with RSV (DeVincenzo et al. 2010) and an accurate approximation using a growing and decaying exponentials (Smith et al. 2010). Both clinically and in the model, higher viral load and delayed viral clearance are independent factors for clinical course in RSV-infected subjects. The viral load is defined as the titer detected in the body using qPCR or plaque assay in many studies (DeVincenzo et al. 2010; Saleeby et al. 2011). Increased viral load is associated with delayed viral clearance, greater disease severity, higher risk of intensive care, prolonged hospitalization, and respiratory failure (DeVincenzo et al. 2010; Saleeby et al. 2011). The severity of symptoms correlates with the initial rise and peak of viral load and decreases with decrease in viral load (DeVincenzo et al. 2010). The infection rate, viral load, and disease severity were unrelated to the initial quantity of RSV (DeVincenzo et al. 2010), implying that individual susceptibility or level of immunity is more important in determining the course of infection. Our model treats viral dynamics as a given response to intrinsic susceptibility or level of immunity. In reality, both the peak and the duration of virus depend on the dynamic immune response, and the model could be extended to include this feedback.
Viral respiratory infections not only exacerbate asthma but are also involved in its initial development. Recurrent severe respiratory virus infections in early life are accepted as an important cause for the development of asthma later in life (Kusel et al. 2007). Viral respiratory infections in the first two years are significantly associated with allergic sensitization (Holt et al. 2010). Airway remodeling, acute inflammation, and host susceptibility interact with each other and with long-term inflammation to generate the structural changes that trigger the eventual development of asthma (Sumi and Hamid 2007). Our model provides a framework for extensions to explain more severe and longer-lasting symptoms, and even exacerbation of asthma caused by respiratory viral infections. We are particularly interested in extending the model to investigate recurrent viral infections, and how an initial damaging infection predisposes a host to later sensitization and the slow progression to asthma.
References
Akaike T, Fujii S, Kato A et al (2000) Viral mutation accelerated by nitric oxide production during infection in vivo. Faseb J 14:1447–1454
Boukhvalova MS, Prince GA, Soroush L, Harrigan DC, Vogel SN, Blanco JC (2006) The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine 24:5027–5035
Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI (1992) Cytokines in symptomatic asthma airways. J Allergy Clin Immunol 89:958–967
Busse WW, Lemanske RF Jr, Gern JE (2010) Role of viral respiratory infections in asthma and asthma exacerbations. Lancet 376:826–834
Chernyavsky IL, Croisier H, Chapman LA et al (2014) The role of inflammation resolution speed in airway smooth muscle mass accumulation in asthma: insight from a theoretical model. PLoS One 9:e90162
Childs LM, Paskow M, Morris SM Jr, Hesse M, Strogatz S (2011) From inflammation to wound healing: using a simple model to understand the functional versatility of murine macrophages. Bull Math Biol 73:2575–2604
Contoli M, Message SD, Laza-Stanca V et al (2006) Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med 12:1023–1026
Corne JM, Marshall C, Smith S et al (2002) Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet 359:831–834
Curran JN, Winter DC, Bouchier-Hayes D (2006) Biological fate and clinical implications of arginine metabolism in tissue healing. Wound Repair Regen 14:376–386
Development Core Team R (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
DeVincenzo JP, Wilkinson T, Vaishnaw A et al (2010) Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med 182:1305–1314
Dolin R (2015) Common viral respiratory infections. In: Kasper DL et al (eds) Harrison’s principles of internal medicine, 19th edn. McGraw-Hill Education, New York, pp 1205–1209
Dougherty RH, Fahy JV (2009) Acute exacerbations of asthma: epidemiology, biology and the exacerbation-prone phenotype. Clin Exp Allergy 39:193–202
Dweik RA, Boggs PB, Erzurum SC et al (2011) An official ATS clinical practice guideline: interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am J Respir Crit Care Med 184:602–615
El Saleeby CM, Bush AJ, Harrison LM, Aitken JA, Devincenzo JP (2011) Respiratory syncytial virus load, viral dynamics, and disease severity in previously healthy naturally infected children. J Infect Dis 204:996–1002
Erdman DD, Weinberg GA, Edwards KM et al (2003) GeneScan reverse transcription-PCR assay for detection of six common respiratory viruses in young children hospitalized with acute respiratory illness. J Clin Microbiol 41:4298–4303
Folcik VA, An GC, Orosz CG (2007) The basic immune simulator: an agent-based model to study the interactions between innate and adaptive immunity. Theor Biol Med Model 4:39
Gehlhar K, Bilitewski C, Reinitz-Rademacher K, Rohde G, Bufe A (2006) Impaired virus-induced interferon-alpha2 release in adult asthmatic patients. Clin Exp Allergy 36:331–337
GINA (2014) Global strategy for asthma management and prevention, global initiative for asthma (GINA) 2014. Available from: http://www.ginasthma.org/. Date last updated. 2014
Haldar P, Pavord ID, Shaw DE et al (2008) Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med 178:218–224
Holt PG, Rowe J, Kusel M et al (2010) Toward improved prediction of risk for atopy and asthma among preschoolers: a prospective cohort study. J Allergy Clin Immunol 125:653–659
Hussell T, Bell TJ (2014) Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol 14:81–93
Hussell T, Spender LC, Georgiou A, O’Garra A, Openshaw PJ (1996) Th1 and Th2 cytokine induction in pulmonary T cells during infection with respiratory syncytial virus. J Gen Virol 77(Pt 10):2447–2455
Iikura K, Katsunuma T, Saika S et al (2011) Peripheral blood mononuclear cells from patients with bronchial asthma show impaired innate immune responses to rhinovirus in vitro. Int Arch Allergy Immunol 155(Suppl 1):27–33
Jackson DJ, Gangnon RE, Evans MD et al (2008) Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med 178:667–672
Jackson DJ, Johnston SL (2010) The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol 125:1178–1187
Jacoby DB (2004) Virus-induced asthma attacks. J Aerosol Med 17:169–173
Jiang H, Harris MB, Rothman P (2000) IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol 105:1063–1070
Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD (1998) Rapid interferon gamma-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J Exp Med 188:1541–1546
Khaitov MR, Laza-Stanca V, Edwards MR et al (2009) Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 64:375–386
Khallou-Laschet J, Varthaman A, Fornasa G et al (2010) Macrophage plasticity in experimental atherosclerosis. PLoS One 5:e8852
Kharitonov SA, Yates D, Barnes PJ (1995) Increased nitric oxide in exhaled air of normal human subjects with upper respiratory tract infections. Eur Respir J 8:295–297
Kim CK, Kim SW, Park CS, Kim BI, Kang H, Koh YY (2003) Bronchoalveolar lavage cytokine profiles in acute asthma and acute bronchiolitis. J Allergy Clin Immunol 112:64–71
Kim Y, Lee S, Kim YS et al (2013) Regulation of Th1/Th2 cells in asthma development: a mathematical model. Math Biosci Eng 10:1095–1133
Kleiner G, Marcuzzi A, Zanin V, Monasta L, Zauli G (2013) Cytokine levels in the serum of healthy subjects. Mediat Inflamm 2013:434010
Kolodziejski PJ, Koo JS, Eissa NT (2004) Regulation of inducible nitric oxide synthase by rapid cellular turnover and cotranslational down-regulation by dimerization inhibitors. Proc Natl Acad Sci USA 101:18141–18146
Kusel MM, de Klerk NH, Kebadze T et al (2007) Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol 119:1105–1110
McKenna SD, Vergilis K, Arulanandam AR, Weiser WY, Nabioullin R, Tepper MA (2004) Formation of human IFN-beta complex with the soluble type I interferon receptor IFNAR-2 leads to enhanced IFN stability, pharmacokinetics, and antitumor activity in xenografted SCID mice. J Interferon Cytokine Res 24:119–129
Modolell M, Corraliza IM, Link F, Soler G, Eichmann K (1995) Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol 25:1101–1104
Moore WC, Meyers DA, Wenzel SE et al (2009) Identification of asthma phenotypes using cluster analysis in the severe asthma research program. Am J Respir Crit Care Med 181:315–323
Murphy J, Summer R, Wilson AA, Kotton DN, Fine A (2008) The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol 38:380–385
Proud D, Chow CW (2006) Role of viral infections in asthma and chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 35:513–518
Rath M, Muller I, Kropf P, Closs EI, Munder M (2014) Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol 5:532
Richardson JY, Ottolini MG, Pletneva L et al (2005) Respiratory syncytial virus (RSV) infection induces cyclooxygenase 2: a potential target for RSV therapy. J Immunol 174:4356–4364
Shaykhiev R, Krause A, Salit J et al (2009) Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 183:2867–2883
Shirey KA, Pletneva LM, Puche AC et al (2010) Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol 3:291–300
Smith AM, Adler FR, Perelson AS (2010) An accurate two-phase approximate solution to an acute viral infection model. J Math Biol 60:711–726
Soetaert K, Petzoldt T, Setzer RW (2010) Solving differential equations in R: package deSolve. J Stat Softw 33:1–25
Song L, Guo Y, Deng Q, Li J (2012) TH17 functional study in severe asthma using agent based model. J Theor Biol 309:29–33
Sturzebecher S, Maibauer R, Heuner A, Beckmann K, Aufdembrinke B (1999) Pharmacodynamic comparison of single doses of IFN-beta1a and IFN-beta1b in healthy volunteers. J Interferon Cytokine Res 19:1257–1264
Sumi Y, Hamid Q (2007) Airway remodeling in asthma. Allergol Int 56:341–348
Syrmis MW, Whiley DM, Thomas M et al (2004) A sensitive, specific, and cost-effective multiplex reverse transcriptase-PCR assay for the detection of seven common respiratory viruses in respiratory samples. J Mol Diagn 6:125–131
Takaoka A, Hayakawa S, Yanai H et al (2003) Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516–523
Tsutsumi H, Takeuchi R, Ohsaki M, Seki K, Chiba S (1999) Respiratory syncytial virus infection of human respiratory epithelial cells enhances inducible nitric oxide synthase gene expression. J Leukoc Biol 66:99–104
van oud Alblas AB, van Furth R (1979) Origin, Kinetics, and characteristics of pulmonary macrophages in the normal steady state. J Exp Med 149:1504–1518
Virchow JC Jr, Walker C, Hafner D et al (1995) T cells and cytokines in bronchoalveolar lavage fluid after segmental allergen provocation in atopic asthma. Am J Respir Crit Care Med 151:960–968
Wark PA, Grissell T, Davies B, See H, Gibson PG (2009) Diversity in the bronchial epithelial cell response to infection with different rhinovirus strains. Respirology 14:180–186
Wark PA, Johnston SL, Bucchieri F et al (2005) Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 201:937–947
Weigl JA, Puppe W, Grondahl B, Schmitt HJ (2000) Epidemiological investigation of nine respiratory pathogens in hospitalized children in Germany using multiplex reverse-transcriptase polymerase chain reaction. Eur J Clin Microbiol Infect Dis 19:336–343
Wink DA, Hines HB, Cheng RY et al (2011) Nitric oxide and redox mechanisms in the immune response. J Leukoc Biol 89:873–891
Acknowledgements
This study was supported by the Soonchunhyang University Research Fund and the 21st century Science Initiative Grant from the James S. McDonnell Foundation (FRA).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Lee, J., Adler, F.R. & Kim, P.S. A Mathematical Model for the Macrophage Response to Respiratory Viral Infection in Normal and Asthmatic Conditions. Bull Math Biol 79, 1979–1998 (2017). https://doi.org/10.1007/s11538-017-0315-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11538-017-0315-0