
Abstract
Atherosclerosis, the leading cause of death in the US, is a disease in which a plaque builds up inside the arteries. The low density lipoprotein (LDL) and high density lipoprotein (HDL) concentrations in the blood are commonly used to predict the risk factor for plaque growth. In a recent paper (Hao and Friedman in Plos One e90497, 2014), we have developed a mathematical model of plaque growth which includes the (LDL, HDL) concentrations. In the present paper, we have refined that model by including the effect of reverse cholesterol transport. By exploration-by-examples of regression of a plaque in mice, our model simulations suggest that such drugs as used for mice may also slow plaque growth in humans. We next proceeded to explore the effects of oxidative stress or antioxidant deficiency, high blood pressure and cigarette smoking as risk factors. We suggest for an individual in one of these three risk categories and with specified (LDL, HDL) concentration, how to reduce or eliminate the risk of atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Atherosclerosis, hardening of the artery, is the leading cause of death in the USA and worldwide. The disease triggers heart attack or stroke, with total annual death of 900,000 in the USA (Hoyert and Xu 2012) and 13 million worldwide (http://www.who.int/mediacentre/factsheets/fs310/en/). The disease originates from a plaque which grows in the artery. The plaque consists of extracellular matrix (ECM), macrophages, foam cells, and smooth muscle cells (SMCs). As the plaque grows, it progressively restricts blood flow, resulting in insufficient blood supply to downstream tissue and cells death. Rupture of the fibrous cap of the plaque can also induce thrombus formation in the lumen (Deepa et al. 2009), which may occlude downstream arteries. Plaque rupture in an artery to the brain results in a stroke, while a coronary thrombus causes myocardial infarction, i.e., a heart attack.
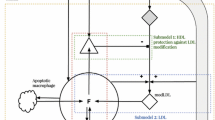
The process of plaque development begins with a lesion in the endothelial layer, enabling LDL and HDL to move from the blood into the intima and become oxidized by free radicals (Cohen et al. 2012; McKay et al. 2005; Ryu 2000); the free radicals are always present in the intima, where they arise from metabolic processes such as ATP production in mitochondria (Cadenas and Davies 2000), and metabolism in human erythrocytes (Cimen 2008). Sensing the presence of the oxidized LDL (ox-LDL) in the intima, endothelial cells begin to secrete monocyte chemoattractant protein (MCP)-1 (Harrington 2000; Reape and Groot 1999), which attracts monocytes into the intima (Osterud and Bjorklid 2003). After entering the intima, monocytes differentiate into macrophages, which endocytose the ox-LDL (Gui et al. 2012; Johnson and Newby 2009; Little et al. 2009). The ingestion of large amounts of ox-LDL transforms the fatty macrophages into foam cells (Calvez et al. 2009; Gui et al. 2012). Smooth muscle cells (SMCs) from the media move into the intima by chemotactic forces due to MCP-1 (Harrington 2000) and platelet-derived growth factor (PDGF) (Reape and Groot 1999; Ross et al. 1990), as well as by haptotaxis from the extracellular matrix (ECM); PDGF is secreted by macrophages, foam cells and SMCs (Raines and Ross 1993; Ross et al. 1990). ECM is remodeled by matrix metalloproteinase (MMP) produced by variety of cells including SMCs (Rodriguez et al. 2007), and is inhibited by tissue inhibitor of metalloproteinase (TIMPs) produced by macrophages and SMCs (Fabunmi et al. 1998). Interleukin IL-12, secreted by macrophages and foam cells (Gui et al. 2012; King and Segal 2005; Reape and Groot 1999), contributes to the growth of a plaque by activating T cells (Hansson et al. 1983; Harrington 2000; King and Segal 2005). The activated T cells secrete interferon IFN-\(\gamma \), which in turn activates macrophage (Hansson et al. 1983; Johnson and Newby 2009; Kosaka et al. 1992) in the intima. Oxidized HDL (ox-HDL) is not ingested by macrophages. Furthermore, HDL removes (oxidized) cholesterol from foam cells and transports them back to the liver, a process called reverse cholesterol transport (RCT) (Alexander et al. 2009; Azzam and Fessler 2012; Cucuianu et al. 2007; Cuchel and Rader 2006; Llera-Moya et al. 2010; Feig et al. 2011a, b; Francis and Pierce 2011; Tall et al. 2001; Vikstedt et al. 2007; Wang et al. 2007a, b; Zhou et al. 2008). The removal of cholesterol by HDL is enabled by proteins such as ABCA1 and ABCG1 (ATP-binding cassette transporters) in the foam cell (Alexander et al. 2009; Azzam and Fessler 2012; Cuchel and Rader 2006; Llera-Moya et al. 2010; Vikstedt et al. 2007; Wang et al. 2007b; Zhou et al. 2008). RTC reduces the inflammation as it converts pro inflammatory plaque macrophages, particularly foam cells, into anti-inflammatory M2 macrophages (Feig et al. 2011b; Moore et al. 2013; Sanson et al. 2013). Intervention schemes, which up-regulate the expression of ABCA1 or ABCG1, can actually result in plaque regression (Cuchel and Rader 2006; Chen et al. 2013; Francis and Pierce 2011; Feig et al. 2011b; Hu et al. 2014; Lovren et al. 2012; Rayner et al. 2011; Schiopu et al. 2007; Wang et al. 2007a). Figure 1 illustrates the geometry within the intima as the process of plaque growth begins, and Fig. 2 is a schematics of the network of cells and cytokines within the plaque. In the present paper, we develop a mathematical model based on the network shown in Fig. 2 and use it to explore risk factors of atherosclerosis in humans.

Schematics of the initiation of a plaque. LDL and HDL cross damaged inner arterial wall into the intima and undergo oxidation. The oxidized LDL (ox-LDL) causes inflammation, which brings in macrophages. The macrophages endocytose the ox-LDL and become foam cells. HDL removes cholesterol from foam cells with protein transporter ABCA1, and foam cells become anti-inflammatory M2 macrophages

Schematic network of atherosclerosis
Plaque buildup in the arteries is a natural part of the aging process. The buildup is progressive. For example, between the ages of 50–60 only 1 % of adults have major plaque in the carotid arteries, while by the ages of 80–90, 10 % of adults have substantial plaque buildup (Ralph et al. 1997). The most important risk factors of plaque buildup are the following:
-
High cholesterol;
-
High blood pressure;
-
Cigarette smoking, and
-
Oxidative stress, or antioxidants deficiency.
Other risk factors include diabetes, obesity and physical inactivity. Indeed, patients with diabetes are more likely to have high blood pressure as well as high cholesterol, both of which increase the risk of atherosclerosis; being overweight contributes to high blood pressure and diabetes, both of which increase the risk of carotid artery disease; and physical inactivity does not provide the benefits of exercise which reduce all the above risk factors. A family history of atherosclerosis is also a risk factor.
-
High cholesterol Very low-density lipoprotein (VLDL) is made by the liver for transport of cholesterol in the blood. VLDL also transports triglycerides formed in the liver. VLDL travel through the blood vessels unloading the fat throughout the body, where VLDL become low-density lipoprotein (LDL). The initiation of a plaque is caused by a lesion in the artery. Pieces of LDL get stuck in the blood vessel wall around the lesion and begin a process of narrowing of the lumen. High density lipoprotein (HDL) clears cholesterol from the blood vessel walls. Thus, the process of plaque formation or elimination depends on the balance between LDL (the “bad” cholesterol) and HDL (the “good” cholesterol).
-
High blood pressure Blood pressure measures the force against the arterial walls. High blood pressure promotes plaque formation by causing damage to arterial walls and by increasing the flux of monocytes into the plaque (American Heart Association 2013). As the plaque continues to grow, the artery becomes narrowed which further restricts blood flow. This causes additional increase in the blood pressure at the plaque location, with a more likely outcome of plaque rupture. A blood pressure of 140/90 is considered high, and can force scarring or lesion on the artery walls.
-
Cigarette smoking Blood vessels have a smooth inner lining that can slow the process of atherosclerosis. Cigarette smoking contributes directly to artery disease by damaging the smooth inner lining of arteries throughout the body (Ambrose and Barua 2004; Powell 1998). This damage diminishes the ability of the arteries to prevent blockages and contributes to the formation of plaque deposits on damaged areas of the arteries. Smoking also increases blood pressure and lowers levels of HDL (He et al. 2013; Neufeld et al. 1997).
-
Oxidative stress, or insufficiency of antioxidants The body produces free radicals. Free radicals are generated largely during the production of ATP in mitochondria (Cadenas and Davies 2000) but also by other biological processes (Cimen 2008). Radicals leak from the mitochondria and form reactive oxygen species (ROS) such as superoxide anions (\(\mathrm{O}_2^-\)) and hydroxyl radicals (OH). These species lead to the production of hydrogen peroxide (\(\mathrm{H}_2\mathrm{O}_2\)) from which a cascade of further hydroxyl radicals are generated. Radicals can be both beneficial and harmful to biological tissues. They play a crucial role in stimulation of phagocytosis, however, over production of radicals forms an oxidative stress which increases the pathogenesis of atherosclerosis. Antioxidants combine with free radicals and neutralize them. A plaque forms after LDL from the blood enters into the intima layer of the blood vessel wall and becomes oxidized by free radicals. Radical scavenging antioxidants inhibit LDL oxidation and thus reduce the risk of atherosclerosis. Hence, antioxidants should have protective and therapeutic effects against atherosclerosis, and increased oxidative stress, or deficiency of antioxidant is an important risk factor to the arterial disease. Numerous in vitro studies show that vitamin E is a radical scavenging antioxidant which inhibits LDL oxidation efficiently, especially together with vitamin C (Cynshi et al. 1998; Noguchi and Niki 1998). However, several human clinical trials of antioxidants vitamin C and vitamin D have failed, so far, to show reduction in cardiovascular events (Moore et al. 2013).
The American College of Cardiology (ACC) and the American Heart Association (AHA) have recently issued the 2013 guidelines for atherosclerosis risk associated with cholesterol levels (Stone et al. 2013). Their new cholesterol guidelines are much stricter than the older guidelines of the Third Adult Treatment Panel (ATP III) by the National Cholesterol Education Program (2002). As pointed out in Pencina et al. (2014), the ACC-AHA guidelines could result in nearly 13 million more adults in the USA being put on statins. As the guidelines for assessing the risk associated with atherosclerosis keep changing from one report to another, it becomes important to develop a mechanistic approach which explains how cholesterol levels actually affect plaque growth which leads heart attack or stroke.
In a recent paper, Hao and Friedman (2014) developed such a mechanistic model of plaque formation. Denoting by \(L_0\) and \(H_0\) the concentrations of LDL and HDL in the blood, their model includes \(L_0\) and \(H_0\) as parameters which determine whether a small plaque will continue to grow or shrink over time. More precisely, given any point \((L_0,H_0)\), they determined the percentage of growth or shrinkage of the plaque over a period of 300 days. Accordingly, they set up a “risk map” for atherosclerosis depending on the combined levels of \(L_0\) and \(H_0\). The predictions of the model agree with the general guidelines of the AHA. The risk map can also be used to suggest which of the available anti-cholesterol drugs would be the best for an individual at risk with specific \(L_0, H_0\) values.
The model in Hao and Friedman (2014), however, did not take into account the RCT process. The present paper refines the mathematical model of Hao and Friedman (2014) by taking account of the RCT process. An ODE model of foam cell formation which includes RCT was recently proposed by Zhang et al. (2013). The simulations of our new model differ only slightly from the simulations in Hao and Friedman (2014), and, in particular, the new risk map (Fig. 14 in Sect. 4) is still in agreement with the AHA guidelines. But the new model enables us to explore intervention methods that achieve plaque regression by up-regulation of ABCA1 (or ABCG1) while keeping \((L_0,H_0)\) fixed. Simulation of such interventions with various drugs are shown to agree with mice experimental results reported in Chen et al. (2013), Feig et al. (2011b), Lovren et al. (2012), Rayner et al. (2011) and Schiopu et al. (2007). This suggests that such drugs may stop plaque growth also in human artery.
The risk map in Fig. 14 does not take into account the last three of the four risk factors mentioned above. However, the model includes two parameters which are related to these risk factors: the concentration of radicals \((r_0)\) in the arterial wall, and the extend of the lesion in the artery, represented by a parameter \(\alpha \). In the Sect. 6, we consider the risk associated with the four parameters \((r_0,\alpha ,L_0,H_0)\). In particular, we simulate the risk associated with \((L_0,H_0)\) for different values of \(r_0\) and \(\alpha \). We show how the \((L_0,H_0)\) risk map is altered by decreasing \(r_0\), that is, by antioxidant enrichment, and how the \((L_0,H_0)\) risk map changes as \(\alpha \) increases as a result of high blood pressure or cigarette smoking. There is an increasing number of drugs to lower the risk of atherosclerosis. In Sect. 6, we explain how to determine which drug would be best for a person with specified values of \((r_0,\alpha ,L_0,H_0)\).
2 The Mathematical Model
The mathematical model is based on the diagram shown in Fig. 2. It is assumed that the plaque has the consistency of a porous medium, so that by Darcy’s law, \(\mathbf {u}=-\nabla \sigma \) where \(\sigma \) is the pressure caused by the moving cells within the plaque. The variables of the model are listed in Table 1.
The mass of a plaque is the integral of \(M_1+M_2+T+S+F+\rho \) taken over the volume of the plaque. We extend the model in Hao and Friedman (2014) by including reverse cholesterol transport (RCT). The extended model is given by the following system of partial differential equations:
We assume that the total density of all the cells plus the concentration of \(\rho \) is constant. We take the constant to be 1 g/cm\(^3\), i.e.,
We assume that all cells are approximately of the same volume and surface area, so that the diffusion coefficients of the all cells have the same coefficient, \(D\). By adding Eqs. (3), (4), (5), (14) and (15) we get
where
Equation (17) gives a relation between \(\rho \) and \(\sigma \). The equation of the density of ECM is given by
Since \(u=-\nabla \sigma ,\) this equation can be written in the form
where
Adding this equation and (17), we get our final equation for \(\sigma \):
The equation for \(\rho \) can then be written as
The system (1)–(20) (except for the new Eq. 4) coincides with the system in Hao and Friedman (2014) if we drop the term \(\displaystyle \frac{k_{AF}}{1+m}\frac{HF}{K_F+F}\) in Eqs. (1) and (5). This term represents the removal rate of cholesterol from foam cells; for simplicity, we do not include removal of cholesterol from proinflammatory plaque macrophages M1. We also assume that as cholesterol is removed, foam cells \(F\) become anti-inflammatory macrophages M2 (Feig et al. 2011b; Moore et al. 2013; Sanson et al. 2013), which accounts for the loss of HDL and foam cells in Eqs. (1) and (5), respectively, and in the gain of M2 macrophages in Eq. (4). The parameter \(m\) represents the level of intervention which results in increase in ABCA1 and ABCG1. With no intervention, cholesterol reverse transport is small, i.e., \(m\) is large; an increase in the number of ABCA1 (or ABCG1) will decrease the value of the parameter \(m\).
We impose the following boundary conditions:
-
Boundary contact with blood, \(\varGamma _I\) We assume Robin boundary conditions on \(\varGamma _I\),
$$\begin{aligned} \frac{\partial X}{\partial \mathbf {n}}~+~\alpha \alpha _X (X-X_0)~=~0, \end{aligned}$$(23)where \(X\) is \(L, H, M_1, M_2\) or \(T\), and no-flux boundary conditions for all other variables, except for \(\rho \),
$$\begin{aligned} \frac{\partial Y}{\partial \mathbf {n}}~=~0, \end{aligned}$$(24)where \(Y\) is \(L_{ox}, r, P, I_\gamma , S, I_\gamma , I_{12}, G, Q, Q_r\) or \(F\). The coefficient \(\alpha _X\) is a constant except for \(M_1\), and \(\alpha _{M_1}(L_{ox},H)=\tilde{\alpha }_{M_1}\frac{L_{ox}}{1+H}\). We assume that M2 macrophages migrate from the plaque to the vascular system, and take \((M_{2})_{0}=0\). In Hao and Friedman (2014), we fixed \(\alpha =1\), but in Sect. 5.2 \(\alpha \) will take also other values.
The free boundary \(\varGamma _I\) is held together by cell-to-cell adhesion forces, so that
$$\begin{aligned} \sigma =\gamma \kappa \quad \hbox { on }\quad \varGamma _I, \end{aligned}$$where \(\kappa \) is the mean curvature of the surface \(\varGamma _I\), and \(\gamma \) is a positive constant. Furthermore, the velocity \(V_n\) of the free boundary \(\varGamma _I\) in the outward normal direction \(\mathbf {n}\) is
$$\begin{aligned} V_n=-\frac{\partial \sigma }{\partial \mathbf {n}}\quad \hbox { on }\quad \varGamma _I. \end{aligned}$$(25) -
Boundary contact with media, \(\varGamma _M\) We assume no-flux boundary conditions on \(\varGamma _M\) for all variables except \(\rho \) and \(S\): For \(S\), we have
$$\begin{aligned} \frac{\partial S}{\partial \mathbf {n}}+\alpha _S(S-S_0)=0\quad \hbox { on }\quad \varGamma _M, \end{aligned}$$(26)where \(\alpha _S=\alpha _S(P,G)=\tilde{\alpha }_S\frac{P+G}{P_0+G_0}\). The boundary conditions of \(\rho \) on \(\varGamma _I\) and \(\varGamma _M\) are determined by \(\rho =1-S-M_1-M_2-F-T\).
The initial conditions do not significantly affect the state of the plaque after 30 days; what affect the state of the plaque are the parameters \(L_0\) and \(H_0\). For simplicity, we take the following initial conditions: \(X=\frac{X_0}{2}\), for \(X=L,H,r,M_1,M_2,T,S,\rho \), and all other variables are zero.
A summary of all the model parameters is given in Tables 2 and 3. Most of the parameters are taken from Hao and Friedman (2014); We also take all the parameters with \(M_1\) (e.g. \(\lambda _{I_{12}M_1}, K_{M_1}\), etc) to be the same as the corresponding parameters with \(M\) in Hao and Friedman (2014), and take \(d_{M_2}=d_{M_1}=d_{M}\). However, since \(M_2\) are macrophages from which \(L_{ox}\) has been “cleaned” by HDL, they can ingest more \(L_{ox}\) than \(M_1\) macrophages; hence we take \(\lambda _{L_{ox}M_2}>\lambda _{L_{ox}M_1}\).
3 Exploring Interventions for Plaque Regression
In this section, we use the model to explore-by-example intervention methods for plaque regression, by comparing model simulations with experimental data. The data were obtained from several mouse experiments. In the first three experiments (Sects. 3.1–3.3), Ldlr-/- mice were first fed western diet for several months in order to develop a plaque in the artery. Their LDL level reached 1,100 mg/dl while their HDL level increased to 120 mg/dl (Francis and Pierce 2011). The mice were then put on chow diet in order to maintain their cholesterol level, and then treated with anti-atherosclerosis drug. We shall describe three different drug treatments and compare simulations of our mathematical model with these experimental results. In our simulations, we begin with the initial data as specified at the end of Sect. 2, and first run the simulation for 30 days with \(L_0=1{,}100\) mg/dl, \(H_0=120\) mg/dl; for the next 30 days (i.e., from day 31 to day 60), we introduce the drug treatment into our model (by modifying the parameters \(m\) in Sects. 3.1–3.3) and show that the model simulations agree qualitatively with the experimental data. In Sect. 3.4, ApoE knockout mice were used and \(S_0\) was modified to get qualitative agreement with experimental data.
3.1 MicroRNA miR-33 Inhibits the Production of ABCA1
We begin with the experimental results in Rayner et al. (2011) who used anti-miR33 to treat Ldlr-/- mice in order to enhance the production of ABCA1 and establish regression of a plaque. To mimic the experiment of Rayner et al. (2011), we represent the anti-miR33 treatment by decreasing \(m\) from \(m=100\) to \(m=0\) (this corresponds to up-regulation of ABCA1). The simulation shown in Figs. 3, 4, 5 and 6 are in qualitatively agreement with the plaque regression measurement reported in Rayner et al. (2011), for plaque weight, \(M_1\) and \(\rho \). All our simulations are done in 2D planes: in a planar cross section of the artery, and in a planer cross section along the length of the artery; in the latter case, we use periodic boundary conditions along the artery. Figures 3, 7 and 12 illustrate the results of such 2D-simulations. The results from the two cross sections are combined to give the simulations shown in the Figs. 4, 5, 6, 8, 9, 10, 11, 13 and 14.

2D cross section of the plaque for different times: treatment with anti-miR33 began at day 31. The top row displays a cross section of the artery, and the bottom row displays a cross section along the artery

Plaque weight comparison: treatment with anti-miR33 began at day 31

M1 macrophage comparison: treatment with anti-miR33 began at day 31

ECM comparison: treatment with anti-miR33 began at day 31

2D cross section of the plaque for different treatments: treatment with antibody began at day 31. The top row displays a cross section of the artery, and the bottom row displays a cross section along the artery

Plaque weight comparison: treatment with antibody began at day 31

M1 macrophage comparison: treatment with antibody began at day 31

MCP-1 comparison: treatment with antibody began at day 31

Foam cell comparison: treatment with TGF-\(\beta \) for 2 days
3.2 Effect of Antibody Treatments
In Schiopu et al. (2007), antibody treatments were used to increase the production of ABCA1 and demonstrate regression of the plaque in mice. Figures 7, 8, 9 and 10 show simulation results which agree qualitatively with the experimental results on plaque weight, M1 and MCP-1 as reported in Schiopu et al. (2007), after adjusting the parameter \(m: m=100, m=80\) and \(m=40\) for different antibody treatments. As in the simulation in Sect. 3.1, the simulation of antibody treatment began at day 31.
3.3 Effect of TGF-\(\beta \)
In Panousis et al. (2011), TGF-\(\beta \) was reported to increase cholesterol efflux in mice, and to increase the levels of ABCA1 and the corresponding decrease in foam cells. We represent the increase in ABCA1 by the TGF-\(\beta \) treatment by decreasing \(m\) from \(m=100\) to \(m=60\) after 30 days. Figure 11 simulates this experiment and shows qualitative agreement with the results in Panousis et al. (2011).
3.4 Effect of miR-145
In Lovren et al. (2012), miR-145 was used to target SMCs, thereby reducing plaque weight. We can model this experiment by decreasing the influx rate of SMCs, which are attracted to the plaque from the media: we represent the miR145 treatment by decreasing \(S_0\) from \(S_0=0.3\) to \(S_0=0\). Figures 12 and 13 agree qualitatively with the experimental results in Lovren et al. (2012).

2D cross section of the plaque for different time: treatment with miR-145 began at day 31. The top row displays a cross section of the artery, and the bottom row displays a cross section along the artery

Plaque weight comparison: treatment with miR-145 began at day 31
4 Risk Map
In Hao and Friedman (2014), we took \(\alpha =1\) and computed a “risk map” for humans, associating with each point \((L_0,H_0)\), the percentage of plaque mass growth or shrinkage at the end of 300 days. In the computations, the initial plaque was taken to be 2D cross section, either orthogonal to the blood vessel or along the blood vessel. Such a map can be recomputed for the extended RCT model.
To create the risk map, we divided the LDL axis by 121 equidistant points \(L^i=70+i\) (\(i=0,\ldots , 120\)) and divided the HDL axis by 21 equidistant points \(H^j=40+j\) (\(j=0,\ldots , 20\)). For each pair \((L^i,H^j)\), we computed the weight of the plaque, \(W(L^i,H^j)\), after 300 days, and formed the risk matrix
where \(W_0\) is the initial weight of the plaque. We then developed a color map (with \(\alpha =1\)) as shown in Fig. 14 which interpolates the risk matrix associated with percentage of plaque growth to each point in the \((L_0,H_0)\) plane. Having formed such a color map, we can then introduce a contour line for any given percentage of plaque growth or shrinkage.

Risk map for \(m=100\)
The risk map is slightly different than the risk map in Hao and Friedman (2014), if m is large, so that the effect of RCT is small when no treatment is applied. The risk associated with \((L_0,H_0)\) here is somewhat lower than in Hao and Friedman (2014), but both risk maps are in agreement with AHA guidelines, which are not very precise.
5 Risk Factors for Human Atherosclerosis
Here, for illustration, we consider just two contours: 0 % growth and 10 % growth. The contour for 0 % growth is a curve:
above which there is no risk of atherosclerosis, and contour for 10 % growth is a curve
below which there is a high risk of atherosclerosis. The region between the two contours is of low risk for atherosclerosis. The two curves in Fig. 14 are \(\varGamma _{r_0,1}^1\) and \(\varGamma _{r_0,1}^2\).
5.1 The Dependence of the Risk Map on \(r_0\)
We first consider the case of a fixed \(\alpha =1\) (as in Fig. 14) and study the effect of decreasing \(r_0\) from the value \(r_0=0.26\) used in Hao and Friedman (2014) to \(r_0=0.2\); this corresponds to antioxidant enrichment by change of diet, supplementary antioxidant pills, and exercise. Figure 15 shows the comparison between \(\varGamma ^i_{0.26,\alpha }\) and \(\varGamma ^i_{0.2,\alpha }\) (\(i=1,2\)). Clearly \(\varGamma ^i_{0.2,\alpha }\) lies below \(\varGamma ^i_{0.26,\alpha }\) for \(i=1,2\). But we can get much more specific information from Fig. 15. Consider, for example, the two individuals A, B depicted in Fig. 15, both are in the high-risk zone if their level of antioxidant is insufficient. By enriching their antioxidant, A will move into the no-risk zone while B will move into the low-risk zone.

Comparison of risk zones for two different values of \(r_0\)
5.2 The Dependence of the Risk Map on \(\alpha \)
We can follow the above procedure with \(r_0\) fixed as in Fig. 14 and \(\alpha \) increased from its value \(\alpha = 1\) in Fig. 14 to \(\alpha =5\); this corresponds to increased damage to an arterial wall as a result of high blood pressure or cigarette smoking. The comparison between \(\varGamma ^i_{r_0,1}\) and \(\varGamma ^i_{r_0,5}\) for \(i=1,2\) are shown in Fig. 16. If high blood pressure individuals D and E can reduce their blood pressure, they will move from the high-risk zone to the no-risk zone for D and low-risk zone for E. Similarly, C can reduce the risk from low-risk zone to no-risk zone.

Comparison of risk zones for two different values of \(\alpha \)
6 Personalized Treatment for Atherosclerosis
As illustrated in Figs. 15 and 16, an individual can change from high-risk zone to low-risk zone, or to no-risk zone by change of life style, e.g. by antioxidant-rich diet including vitamin C and E, or by quit smoking; reducing blood pressure may require medication in addition to a change of life style. But, in addition to change in life style, an individual in a high-risk zone may also need to take anti-cholesterol medication. Our model may suggest a combined therapy which includes, for example, decreasing \(r_0\) by antioxidant, and anti-cholesterol medication which decreases \(L_0\) and increases \(H_0\). Individual A in Fig. 15 can move to the no-risk zone just by an enrichment of antioxidants, with no need to take anti-cholesterol drugs. On the other hand, individual B in Fig. 15 cannot get into the no-risk zone by antioxidant enrichment alone, he/she needs to take also anti-cholesterol drug.
As seen in Fig. 16, individuals D and E can move from the high-risk zone by reducing blood pressure (or quit smoking), but whereas D will move into the no-risk zone, E will only progress to the low-risk zone and will still need to take anti-cholesterol drug. Similarly, C can move from low-risk zone to no-risk zone either by reducing blood pressure (or quit smoking) or by taking anti-cholesterol drug. Some anti-cholesterol drugs, such as statins, lower LDL and at the same time also increase the HDL. Other drugs just decrease the LDL or just increase the HDL. As shown in Hao and Friedman (2014), the choice of anti-cholesterol drug depends on the location of \((L_0,H_0)\) in the risk map, more precisely, on the distance to the no-risk zone. We hypothesize that, barring side effects, the best drug is the one which brings \((L_0,H_0)\) to the nearest point \((\bar{L}_0,\bar{H}_0)\) on the 0 % contour, i.e., to the nearest point on boundary of the no-risk zone. Thus, for instance, in the case of individual A in Fig. 15, the best drug is one that mostly increase HDL.
7 Conclusion
Atherosclerosis, hardening of the artery, is the leading cause of death in the United States and worldwide. The disease originates from a plaque grown in the artery. Whereas plaque buildup is a natural part of the aging process, there are specific risk factors that can be reduced by a change in life style. The concentration of LDL and HDL have long been considered as biomarkers for assessing the risk of atherosclerosis. Here, we extended the model developed in Hao and Friedman (2014) to include RCT and used the model to explore-by-example various treatments that demonstrated plaque regression in mice. We have shown that, under these drug treatments, our model for human atherosclerosis yields results that are in qualitative agreement with the mice experiments for atherosclerosis regression. Currently, there exist no drugs that reverses or just stops atherosclerosis in humans; the simulations of our model suggest that drugs explored in Sect. 3 may be effective in stopping the growth of a plaque in human artery, once the plaque has been detected.
We next created a color risk map in the \((L_0,H_0)\)-plane, so that each contour corresponds to a percentage of growth or shrinkage of a small initial plaque over a period of 300 days. The risk map depends on the parameters of the model.
We focused on two important parameters, \(r_0\) and \(\alpha \): for an individual on antioxidant-rich diet \(r_0\) will be decreased; for an individual with a high blood pressure, or for a heavy cigarette smoker, by reducing the blood pressure or by quit smoking, the parameter \(\alpha \) will be reduced. We have shown, with the aid of the risk map, how the reduction of these two parameters, \(r_0\) and \(\alpha \), will lower the risk of atherosclerosis. Although our results are consistent with American Heart Association guidelines, and qualitatively agree with experimental results for mice, in the lack of sufficient human experimental data, our conclusions can only be viewed as qualitative. Furthermore, our model does not include triglycerides (Arsenault et al. 2009), and does not differentiate between LDL cholesterol patterns A and B: only the later pattern is associated with increasing risk of atherosclerosis (http://www.health.am/ 2004). As more data become available on the effects of triglycerides and LDL cholesterol patterns A and B within a plaque, our model could then be extended to include these additional variables.
We have not considered in this paper the age factor: plaque buildup is increased with age (Ryu 2000). The age factor, however, could be incorporated into our model if we simply associate a large initial plaque to an old age group.
References
Alexander ET, Weibel GL, Joshi MR, Vedhachalam C, de la Llera-Moya M, Rothblat GH, Phillips MC, Rader DJ (2009) Macrophage reverse cholesterol transport in mice expressing ApoA-I Milano. Arterioscler Thromb Vasc Biol. 29(10):1496–501
Ambrose JA, Barua R (2004) The pathophysiology of cigarette smoking and cardiovascular disease: an update. Am Coll Cardiol 43:1731–1737
American Heart Association (2013) Heart and artery damage and high blood pressure. http://www.heart.org/Heart-and-Artery-Damage-and-High-Blood-Pressure-UCM-301823-Article.jsp
Arsenault B, Rana J, Stroes S, Despres P, Shah P (2009) Beyond low-density lipoprotein cholesterol: respective contributions of non-high-density lipoprotein cholesterol levels, triglycerides, and the total cholesterol/high-density lipoprotein cholesterol ratio to coronary heart disease risk in apparently healthy men and women. J Am Coll Cardiol 55:35–41
Azzam KM, Fessler MB (2012) Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol Metab 23(4):169–178
Barter P (2005) The role of HDL-cholesterol in preventing atherosclerotic disease. Eur Heart J Suppl 7:4–8
Cadenas E, Davies K (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29:222–230
Calvez V, Ebde A, Meunier N, Raoult A (2009) Mathematical modelling of the atherosclerotic plaque formation. CEMRACS 2008 Modell Numer Simul Complex Fluids 28:1–12
Chen WJ, Zhang M, Zhao GJ, Fu Y, Zhang DW, Zhu HB, Tang CK (2013) MicroRNA-33 in atherosclerosis etiology and pathophysiology. Atherosclerosis 227(2):201–8
Cimen M (2008) Free radical metabolism in human erythrocytes. Clin Chim Acta 390:1–11
Cohen A, Myerscough M, Thompson R (2012) Athero-protective effects of high density lipoproteins (HDL): an ODE model of the early stages of atherosclerosis, preprint
Cuchel M, Rader DJ (2006) Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation 113(21):2548–2555
Cucuianu M, Coca M, Hancu N (2007) Reverse cholesterol transport and atherosclerosis. A mini review. Rom J Intern Med 45(1):17–27
Cynshi O, Kawabe Y, Tsukasa N (1998) Antiatherogenic effects of the antioxidant BO-653 in three different animal models Osamu. Proc Natl Acad Sci USA 95(17):10123–10128
Deepa T, Binu L, Sukesh A (2009) Modelling blood flow and analysis of atherosclerotic plaque rupture under G-force. Bioinform Biomed 23:1–4
de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH (2010) The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol 30(4):796–801
Fabunmi R, Sukhova G, Sugiyama S, Libby P (1998) Expression of tissue inhibitor of metalloproteinases-3 in human atheroma and regulation in lesion-associated cells a potential protective mechanism in plaque stability. Circ Res 83:270–278
Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, Young SG, Fisher EA (2011a) Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 123(9):989–998
Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA (2011b) HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci 108(17):7166–7171
Francis AA, Pierce GN (2011) An integrated approach for the mechanisms responsible for atherosclerotic plaque regression. Exp Clin Cardiol 16(3):77C86
Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y (2012) Diverse roles of macrophages in atherosclerosis: from inflammatory biology to biomarker discovery. Mediat Inflamm 2012:693083
Hansson G, Holm J, Jonasson L (1983) Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol 135:169–175
Hao W, Friedman A (2014) The LDL-HDL profile determines the risk of atherosclerosis: a mathematical model. Plos One e90497
Harrington J (2000) The role of MCP-1 in atherosclerosis. Stem Cells 18:65–66
He B, Zhao S, Peng Z (2013) Effects of cigarette smoking on HDL quantity and function: implications for atherosclerosis. J Cell Biochem 114:2431–2436
Hoyert D, Xu J (2012) Deaths: preliminary data for 2011. National vital statistics reports 61
http://www.health.am/ (2004) What are LDL cholesterol particle size patterns A and B?
Hu YW, Hu YR, Zhao JY, Li SF, Ma X, Wu SG, Lu JB, Qiu YR, Sha YH, Wang YC, Gao JJ, Zheng L, Wang Q (2014) An agomir of miR-144-3p accelerates plaque formation through impairing reverse cholesterol transport and promoting pro-inflammatory cytokine production. PLoS One 9(4):e94997
Johnson J, Newby A (2009) Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol 20:370–378
King I, Segal B (2005) Cutting edge: IL-12 induces CD4+CD25- T cell activation in the presence of T regulatory cells. J Immunol 175:641–645
Kosaka C, Masuda J, Shimokado K, Zen K, Yokota T, Sasaguri T, Ogata J (1992) Interferon-gamma suppresses PDGF production from THP-1 cells and blood monocyte-derived macrophages. Atherosclerosis 97:75–87
Little M, Gola A, Tzoulaki I (2009) A model of cardiovascular disease giving a plausible mechanism for the effect of fractionated low-dose Ionizing radiation exposure. PLoS Computat Biol 5:e1000539
Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, Steer BM, Ingram AJ, Gupta M, Al-Omran M, Teoh H, Marsden PA, Verma S (2012) MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation 126(11 Suppl 1):S81–90
McKay C, McKee S, Mottram N, Mulholland T Wilson S (2005) Towards a model of atherosclerosis. Strathclyde Mathematics Research Report
Moore KJ, Sheedy FJ, Fisher EA (2013) Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13(10):709–721
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) (2002) Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 106:3143–3421
Neufeld E, Mietus-Snyder M, Beiser A, Baker A, Newburger J (1997) Passive cigarette smoking and reduced HDL cholesterol levels in children with high-risk lipid profiles. Circulation 96:1403–1407
Noguchi N, Niki E (1998) Dynamics of vitamin E action against LDL oxidation. Free Radic Res 28(6):561–572
Osterud B, Bjorklid E (2003) Role of monocytes in atherogenesis. Physiol Rev 83:1069–1112
Panousis CG, Evans G, Zuckerman SH (2011) TGF-beta increases cholesterol efflux and ABC-1 expression in macrophage-derived foam cells: opposing the effects of IFN-gamma. J Lipid Res 42(5):856–863
Pencina M, Navar-Boggan M, D’Agostino R (2014) Application of new cholesterol guidelines to a population-based sample. N Engl J Med
Powell J (1998) Vascular damage from smoking: disease mechanisms at the arterial wall. Vasc Med 3:21–28
Raines E, Ross R (1993) Smooth muscle cells and the pathogenesis of the lesions of atherosclerosis. Br Heart J 69:30–37
Ralph P, Sacco L, Benjamin E (1997) Risk factors. Stroke 28:1507–1517
Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ (2011) Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest 121(7):2921–2931
Reape T, Groot P (1999) Chemokines and atherosclerosis. Atherosclerosis 147:213–225
Rodriguez J, Orbe J, Paramo J (2007) Metalloproteases, vascular remodeling, and atherothrombotic syndromes. Rev Esp Cardiol 60:959–967
Ross R, Masuda J, Raines E, Gown A, Katsuda S, Sasahara M, Malden L, Masuko H, Sato H (1990) Localization of PDGF-B protein in macrophages in all phases of atherogenesis. Science 248:1009–1012
Ryu B (2000) Low density lipoprotein (LDL), atherosclerosis and antioxidants. Biotechnol Bioprocess Eng 5:313–319
Sanson M, Distel E, Fisher EA (2013) HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS One 8(8):e74676
Schiopu A, Frendeus B, Jansson B, Soderberg I, Ljungcrantz I, Araya Z, Shah PK, Carlsson R, Nilsson J, Fredrikson GN (2007) Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(-/-)/low-density lipoprotein receptor(-/-) mice. J Am Coll Cardiol 50(24):2313–2318
Stone N, Robinson J, Lichtenstein H, Bairey M, Lloyd-Jones D (2013) 2013 ACC/AHA Guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol
Tall AR, Wang N, Mucksavage P (2001) Is it time to modify the reverse cholesterol transport model? J Clin Invest 108(9):1273–1275
The top 10 causes of death. http://www.who.int/mediacentre/factsheets/fs310/en/. Accessed Dec 18, 2013
Vikstedt R, Metso J, Hakala J, Olkkonen VM, Ehnholm C, Jauhiainen M (2007) Cholesterol efflux from macrophage foam cells is enhanced by active phospholipid transfer protein through generation of two types of acceptor particles. Biochemistry 46(42):11979–11986
Wang MD, Franklin V, Marcel YL (2007a) In vivo reverse cholesterol transport from macrophages lacking ABCA1 expression is impaired. Arterioscler Thromb Vasc Biol 27(8):1837–1842
Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ (2007b) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117(8):2216–2224
Zhang S, Ritter L, Ibragimov A (2013) Foam cell formation in atherosclerosis: HDL and macrophage reverse cholesterol transport. Discrete Contin Dyn Syst 825–835
Zhou X, He W, Huang Z, Gotto AM Jr, Hajjar DP, Han J (2008) Genetic deletion of low density lipoprotein receptor impairs sterol-induced mouse macrophage ABCA1 expression. A new SREBP1-dependent mechanism. J Biol Chem 283(4):2129–2138
Acknowledgments
This research has been supported by the Mathematical Biosciences Institute and the National Science Foundation under Grant DMS 0931642. We would like to express our thanks to Drs. Sanjay Rajagopalan and Andrei Maiseyeu at University of Maryland, Baltimore for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Friedman, A., Hao, W. A Mathematical Model of Atherosclerosis with Reverse Cholesterol Transport and Associated Risk Factors. Bull Math Biol 77, 758–781 (2015). https://doi.org/10.1007/s11538-014-0010-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11538-014-0010-3