
Abstract
Immune checkpoints inhibitors (ICIs) have been a breakthrough, with unique response and survival patterns compared with chemotherapy for patients with advanced Mismatch Repair-deficient/Microsatellite instable (dMMR/MSI) colorectal cancer, but have shown disappointing results in Mismatch Repair-proficient/Microsatellite stable (pMMR/MSS) colorectal cancer. As up to 50% of patients harboring dMMR/MSI advanced cancers will ultimately progress after PD-1 blockade, biomarkers are needed to predict response/resistance to immunotherapy and to select patients for immunomodulating combination therapies. Patients with pMMR/MSS colorectal cancer present with distinct immune profiles compared to dMMR/MSI tumors, giving evidence of different immune escape mechanisms, which could be overcome through individualized immunotherapeutic strategies. In this review we discuss the latest developments in the field of immunotherapy for dMMR/MSI and pMMR/MSS colorectal cancers, and unresolved questions and considerations concerning the use of ICI therapies in this population. Future immunomodulation strategies based on biomarker selection (tumor mutational burden, Immunoscore®, mutational profile) are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Despite dramatic responses to immune checkpoint inhibitors in patients with MSI (microsatellite instability) colorectal cancers (CRCs), nearly half of the patients will ultimately progress and no biomarker is currently validated to correctly predict the resistance or benefit derived from immunotherapy. |
POLE exonucleasic domain mutations are a major alternative biomarker to select the best candidates for immunotherapy in MSS (microsatellite stable) CRCs. |
The incorporation of immuno-genomic analyses into future studies that assess immunomodulation/targeted therapeutic combinations would improve the identification of subsets of patients who benefit from these treatments as well as provide support to move the field towards individualized immunotherapeutic strategies in CRCs. |
1 Introduction
Immune checkpoint inhibitors (ICIs) represent one of the major therapeutic breakthroughs in the history of medical oncology. In this context, metastatic colorectal cancer (mCRC) represent an intriguing entity, with a minority (4–5%) of tumors being highly sensitive to these compounds (i.e., CRC harboring microsatellite instability (MSI) and/or mismatch-repair deficiency (dMMR) [1]), while a vast majority of immunologically “cold” tumors are refractory to immunotherapeutic strategies [2,3,4]. It has been known for years that MSI/dMMR tumors are highly infiltrated by immune cells [5]. These tumors are characterized by a high tumor mutational burden (TMB) with highly immunogenic neoantigens arising from frameshift mutations [6]. Moreover, they are associated with an upregulation of checkpoint inhibitors that exhaust intratumoral cytotoxic T lymphocytes and consequently protect MSI cancer cells from their hostile immune microenvironment [7, 8].
Following the durable complete response of one patient with an MSI/dMMR mCRC treated with anti-PD1 ICI in a phase I study that included various types of unselected solid tumors, research and development of ICIs for CRC focused on MSI/dMMR mCRC [9]. The impressive results of several phase II studies demonstrated that immune checkpoint inhibition is a breakthrough therapeutic strategy for dMMR/MSI mCRC [1, 10, 11]. Pembrolizumab or nivolumab alone or in combination with ipilimumab have since been approved by the US Food and Drug Administration (FDA) for the treatment of chemoresistant MSI/dMMR mCRC patients. Moreover, the FDA has granted accelerated approval to pembrolizumab for advanced MSI/dMMR tumors progressing following prior treatment, in a tissue/site-agnostic fashion [12]. The efficacy of ICIs in MSI/dMMR mCRC is now well established, and trials are currently ongoing to assess ICI efficacy in first-line, adjuvant, or even neoadjuvant settings.
However, questions remain concerning the role of ICIs for the treatment of microsatellite stable (MSS) and mismatch repair-proficient (pMMR) CRC. The combination of ICIs with other anticancer drugs is currently being evaluated in MSS/pMMR mCRC. However, the disappointing results of the phase III IMblaze 370 trial (atezolizumab with or without cobimetinib vs. regorafenib) raises concerns regarding the testing of therapeutic ICI strategies without a guiding biomarker in these “cold” tumors [13]. It has been shown that tumor mutational burden (TMB) could be a biomarker for ICI efficacy in various histologies, and some MSS/pMMR CRCs show a high TMB, notably due to mutations within genes coding for DNA polymerases [14, 15]. Furthermore, the consensus molecular subtype (CMS) classification, which allows for better comprehension of CRC molecular heterogeneity, might be a tool worth considering for the development of new immunotherapeutic strategies in MSS/pMMR CRC [16].
In this review, we highlight how MSI/dMMR has emerged as a powerful biomarker for ICI efficacy in mCRC. Then we focus on the currently explored ways to expand the use of ICIs in CRC beyond MSI/dMMR and also in adjuvant and neoadjuvant settings.
2 Immune Checkpoint Blockade in Colorectal Cancer: From Disappointment to Microsatellite Instability
The evaluation of ICIs in CRCs had initially been disappointing. The results of phase I basket trials including molecularly unselected mCRC patients did not show evidence of clinical activity. Notably, no objective responses were observed amongst the 3, 19, and 18 mCRC patients treated with pembrolizumab, nivolumab (anti PD-1 monoclonal antibodies), or BMS-936559 (anti-PD-L1 monoclonal antibody), respectively [2,3,4].
2.1 Clinical Activity of Immune Checkpoint Inhibitors (ICIs) in Patients with Microsatellite Instability (MSI) and/or Mismatch-Repair Deficiency (dMMR) Colorectal Cancer (CRC)
The breakthrough of immunotherapy in CRC came from one out of the 14 mCRC patients included in the phase I CA209-001 basket trial (NCT00441337) who experienced a durable complete response with nivolumab. This patient’s tumor presented an MSI phenotype [9].
The proof-of-concept phase II study (MK-3475-016, NCT01876511) was designed to assess the efficacy of pembrolizumab in three cohorts of chemoresistant metastatic cancer patients: MSI/dMMR mCRC, MSI/dMMR non-CRC, and MSS/pMMR mCRC [1]. Preliminary results published in the New England Journal of Medicine in 2015 showed a 40%, 57%, and 0% objective response rate (ORR), respectively, in the three pembrolizumab-treated cohorts (N = 11, N = 9, and N = 21). This was the first step to recognize MSI/dMMR as a major predictive tissue-agnostic biomarker for the efficacy of ICIs [12]. The updated data after 28 MSI/dMMR mCRC patients treated with pembrolizumab confirmed these encouraging results, with 11% of complete responses, 46% of partial responses, and only 4% of progressive diseases [17].
Clinical trial results of immune checkpoint blockade in MSI/dMMR mCRC patients are summarized in Table 1. In the Keynote-164 study, pembrolizumab was evaluated in two distinct cohorts: (1) cohort A, after two or more prior lines of therapy including fluoropyrimidine, oxaliplatin, and irinotecan, and; (2) cohort B, after one or more prior line of therapy. In these two cohorts, pembrolizumab was associated with a disease control rate (DCR) of 51% and 44%, and 1-year progression-free survival (PFS) rate of 34% and 41%, respectively [18,19,20].
Nivolumab has also been evaluated in the context of MSI/dMMR mCRC. In the CheckMate-142 phase II trial, nivolumab alone (nivolumab 3 mg/kg every 2 weeks) and its combination with ipilimumab (nivolumab 3 mg/kg every 3 weeks + ipilimumab 1 mg/kg every 3 weeks for four cycles then nivolumab 3 mg/kg every 2 weeks) until disease progression exhibited a 34% and 58% ORR, respectively, with 36% and 12% RECIST1.1 progressive disease at first evaluation, respectively. The 2-year PFS and overall survival (OS) rates were 60% and 74%, respectively, with the combination of nivolumab and ipilimumab [21, 22]. In both cohorts, patients were resistant or intolerant to at least one prior line of chemotherapy before inclusion: 54% and 40% of patients, respectively, received three or more lines of systemic treatment before ICI. In a third cohort of the CheckMate-142 trial, 45 patients with no prior treatment for MSI/dMMR mCRC were treated with nivolumab 3 mg/kg every 2 weeks plus low-dose ipilimumab every 6 weeks until disease progression. ORR was 77% and only one progressive disease was observed; 1-year PFS and OS rates were 77% and 83%, respectively [23].
Durvalumab, an anti-PD-L1 monoclonal antibody, has also been tested in the context of MSI/dMMR mCRC. Amongst 36 MSI/dMMR mCRC patients treated with durvalumab 10 mg/kg IV every 2 weeks (NCT01693562 and NCT02227667.), ORR was 22%, DCR for more than 24 weeks was 47%, and the 2-year OS rate was 54% [24]. Interestingly, patients were treated for a maximum of 12 months whereas in other trials ICI treatment was maintained for 2 years or until disease progression.
2.2 ICIs Compared with Conventional Chemotherapy
Based on the positive results from these clinical trials [1, 10,11,12], pembrolizumab as well as nivolumab alone or in combination with ipilimumab have been granted accelerated approval by the FDA for chemoresistant MSI/dMMR mCRC patients. However, the European Medicine Agency has not yet approved ICIs for this subset of patients.
Several randomized trials are currently evaluating the efficacy of ICIs as compared to standard-of-care chemotherapy ± targeted therapy in a first- or second-line metastatic setting (Table 2). Notably, the KEYNOTE-177 and COMMIT phase III studies might lead to the approval of pembrolizumab and atezolizumab, respectively, in frontline MSI/dMMR mCRC if they meet their primary endpoint. The ongoing randomized phase II trial PRODIGE 54 – SAMCO (NCT03186326) is evaluating the efficacy of avelumab in second-line versus standard-of-care treatment (chemotherapy ± targeted therapy). In the randomized phase IIIb CA209-8HW study (NCT04008030), patients are randomized to receive nivolumab alone (arm A), nivolumab plus ipilimumab (arm B) or investigator’s choice chemotherapy ± targeted therapy (arm C) depending on the number of prior lines (0 or 1 prior line: A versus B versus C; > 1 prior line: A versus B). The main objective is to compare the clinical benefit, as measured by PFS, ORR, and OS, achieved by nivolumab in combination with ipilimumab or by nivolumab monotherapy in patients with MSI/dMMR mCRC.
Importantly, two phase III trials are currently assessing the efficacy of ICIs in the adjuvant setting. The Alliance A021502 trial (NCT02912559) is evaluating mFOLFOX6 ± atezolizumab in patients with stage III colon cancer. In the experimental arm, patients receive 6 months of adjuvant mFOLFOX6 concurrently with atezolizumab, followed by six additional months of atezolizumab monotherapy. The POLEM trial (EudraCT 2017000370_10) is testing the benefit of adding ICIs after the completion of capecitabine-based adjuvant chemotherapy (3 months of capecitabine plus oxaliplatin or 6 months of capecitabine monotherapy) in MSI/dMMR or POLE-mutated stage III CRC. At the end of adjuvant chemotherapy, patients who are randomized to the investigational arm will receive an additional 24 weeks of treatment with avelumab. The primary endpoint is disease-free survival in both trials.
2.3 Biomarkers Within the MSI/dMMR Population
Potential biomarkers of ICI efficacy amongst the MSI/dMMR population are summarized in Table 3. Despite high rates of response and durable clinical benefit with ICIs, 10–50% of MSI/dMMR mCRC tumors exhibit primary resistance to immunotherapy. However, a significant amount of these refractory tumors are mistakenly diagnosed as MSI/dMMR. In a cohort study of 38 mCRC patients included in ICI clinical trials based on a positive MSI and/or dMMR status as determined by local laboratories, misdiagnosis of MSI or dMMR status was responsible for three of five cases of primary resistance to ICI [25]. Thus, the first concern when observing immediate tumor progression under ICIs is the possibility of a misdiagnosed MSI phenotype that requires confirmatory analysis by immunohistochemistry or molecular assay, the alternate hypothesis being tumor pseudo-progression [25, 26].
Preliminary results on potential biomarkers for the efficacy of ICIs amongst MSI/dMMR mCRC did not find significant predictive impact by either RAS/RAF mutational status, tumor PD-L1 expression, the inherited (i.e., Lynch syndrome) or sporadic origin of MMR deficiency, or the tumor mutational load [1, 10, 11, 27]. Concerning the mechanism underlying MMR deficiency, it is worth noting that the results remain questionable since the definition of Lynch syndrome-related cancers and sporadic cases is partly equivocal. In the analysis of ICI clinical trials, MMR deficiency origin was determined by investigators based on past medical history collected from clinical records without a systematic molecular approach that might skew the results [1, 11, 28]. Moreover, it has been recently proven that Lynch-like tumors (MSI tumors without MLH1 promotor hypermethylation, BRAF mutation, or germline MMR gene mutation) arise from biallelic somatic MMR gene inactivation in approximately 50% of patients [29]. Therefore, further studies are warranted to confirm these findings.
Several potential mechanisms of resistance are under evaluation, notably such as deleterious mutations of JAK (found in approximately 10–20% of MSI/dMMR tumors), loss of major histocompatibility complex (MHC) molecules, or B2M (beta-2-microglobulin) loss-of-function mutations (20–30% of MSI CRC) [30,31,32,33]. The latter was not found to be associated with ICI resistance in a cohort of 13 B2M-mutated MSI/dMMR mCRC with ORR of 85% (11/13). Remarkably, the authors showed that MHC class I expression is variable in B2M-mutated MSI CRC [34] but not associated with response to ICIs. Note there are currently no available data on the impact of gut microbiota in MSI/dMMR cancer patients treated with ICIs [35, 36]. Histopathological characteristics such as the amount of extracellular mucin and PD-L1 expression at the invasive front might be associated with resistance to ICIs [37].
The impact of tumor mutational load remains controversial within the MSI/dMMR population. Important recently published works detected a positive correlation between tumor mutational load and the efficacy of ICIs amongst the MSI/dMMR population [38, 39]. However, the sample sizes remain small, with potential tumors misdiagnosed as MSI/dMMR amongst ICI-resistant cases [25]. Moreover, the threshold of tumor mutational load to be considered as a predictive biomarker may be different from other tumor types. Larger translational studies are therefore warranted to establish the role of TMB in MSI/dMMR tumors. Finally, intra- and inter-tumoral heterogeneity might negatively influence ICI efficacy on sporadic MSI/dMMR tumors especially in cases of mixed dMMR/pMMR tumors [40, 41].
The quality of the neoantigens seems essential, as demonstrated in MSI/dMMR tumors treated with ICIs [12]. Even in the context of high TMB and predicted neoantigens, very few effective immune responses directed against tumor-associated neoantigens are detected, suggesting that immune response to ICI may rely only on a few highly immunogenic antigens. These antigens are derived mostly from insertion/deletion mutations (Indels), a hallmark of dMMR tumors [38, 42]. Importantly, this phenomenon may be of particular importance in MSS/pMMR tumors where Indels are rarely detected.
2.4 Controversies in the Clinical Management of MSI/dMMR mCRC Patients Treated with ICIs
Pseudo-progression is a noted phenomenon of ICI therapy. Pseudo-progression is an initial radiographic tumor enlargement (consistent with progression per Response Evaluation Criteria in Solid Tumors criteria (RECIST)) followed by measurable tumor regression, sometimes months to years after therapy initiation. The frequency of pseudo-progression in the MSI/dMMR population is currently unknown but it may be common in this highly sensitive population. Amongst patients from the CheckMate-142 trial with progression of disease as best response (N = 14) and who continued treatment beyond progression (N = 11), those with a reduction (N = 3) or stabilization of target lesions (N = 3) were more likely to survive more than 12 months [21]. Distinguishing pseudo-progression from true disease progression is tricky, and irRECIST (Immune-related RECIST) or iRECIST (immune RECIST) criteria can be helpful [43]. Clinical parameters such as the improvement of performance status, improvement of symptoms, or decrease of serum tumor marker levels may help in the clinician’s decision to continue the ICI and wait for later radiologic evaluations. It is noteworthy that the KEYNOTE-016 trial used irRECIST to assess response, while others such as CheckMate-142 and KEYNOTE-164 used RECIST v.1.1. This difference in radiologic evaluation criteria might explain the difference in ORR across studies [10, 11, 17, 18, 20].
Concerning secondary resections of primary tumor or metastasis after ICI treatment, the data are scarce. Some case reports suggested that pre-operative immunotherapy followed by resection of primary tumor and/or metastatic sites is associated with high rates of pathological complete response, despite the presence of residual lesions on radiological exams [44]. This brings to light some issues in the therapeutic management of ICI-treated patients with MSI/dMMR cancer: (1) what is the best therapeutic strategy for long-term responding patients: ICI continuation, surgical removal of residual lesions, or ICI discontinuation without surgery? and (2) would patients who experience disease progression confined to a single site benefit from surgery? These issues are currently emerging since the number of ICI-treated MSI/dMMR mCRC patients is growing exponentially.
Another emerging crucial concern is the optimal duration of treatment for patients who respond to ICIs. The strategy varies across trials, with a fixed duration of ICI treatment (usually limited to 1 or 2 years of treatment) or the continuation of treatment until progression or unacceptable toxicity, which is a burden for patients and healthcare systems. The question remains unanswered and clinical trials are needed to find the optimal treatment duration.
3 Immunotherapy in Colorectal Cancer Beyond MSI/dMMR
Ongoing clinical trials evaluating therapeutic strategies with ICIs for MSS/pMMR CRC patients are displayed in Table 4.
3.1 Hypermutability Amongst Microsatellite-Stable Colorectal Cancer: DNA Polymerase Mutations
The biological substratum of ICI efficacy on MSI/dMMR tumors relies on immunogenic frameshift mutations and high TMB. Several studies have shown that MSI/dMMR CRC could be detected through TMB evaluation with similar accuracy to polymerase chain reaction or immunohistochemistry [45, 46]. Moreover, there is a high-TMB population who do not exhibit MSI/dMMR but displays an ultramutated phenotype with significantly higher TMB than MSI/dMMR CRC: all these tumors harbor mutations in DNA polymerase genes (Fig. 1) [15, 45, 47].

Association of Polymerase E mutations with microsatellite instable (MSI) status and hypermutability in colorectal cancer (data from the The Cancer Genome Atlas)
In the TCGA cohort (N = 276), 16% of CRC samples (N = 44) exhibited hypermutation (defined as a TMB greater than 12 mutations per 106 bases). Among these, all MSS tumors (N = 7) harbored exonucleasic proofreading domain POLE-mutations (DNA epsilon polymerase) and presented the highest TMB. Similarly, in a study by Stadler and colleagues, 31 out of 224 CRC samples were hypermutated, of which three MSS POLE-mutated tumors presented a TMB threefold higher than MSI/dMMR cases [45]. In a study by Vanderwalde and colleagues, 93 out of 1395 CRC (6.7%) samples were hypermutated (TMB high if ≥ 17 mutations per megabase (mt/mb)) and 80 (5.7%) were MSI (and only four MSI tumors had low TMB) [46].
DNA polymerase enzymes are involved in DNA synthesis and repair during the S phase of the cell cycle. In humans, delta and epsilon polymerases (POLD and POLE) are the main DNA polymerases with a proofreading exonuclease domain activity allowing error correction during replication. Deleterious mutations in the exonuclease domain of POLD and POLE genes are responsible for a dramatic genetic instability associated with an ultramutated tumor phenotype. Germline mutations of POLD and POLE genes are associated with an autosomic dominant familial cancer syndrome favoring notably endometrial cancer and CRC occurrence (PPAP: polymerase proofreading-associated polyposis). Somatic POLE gene mutations may also occur [48, 49]. Interestingly, POLE mutations have been described in MSI/dMMR cancers, but arise distantly to the exonuclease domain and are therefore considered as passenger mutations due to the high genetic instability of MSI/dMMR tumors. This hypothesis is supported by the higher TMB of POLE-mutated MSS CRC compared to POLE-mutated MSI CRC tumors [42, 50].
Exonucleasic domain POLE-mutated CRCs typically arise from the left colon and rectum in younger male patients. Not all POLE mutations seem to be driving hypermutagenesis and the high mutation load phenotype seems restricted to specific hotspots. These mutations are observed in 1% of stage II and III tumors and are associated with favorable prognosis. This population is very rare in the metastatic setting, accounting for less than 1% of cases [47, 51], and prognosis and response to conventional treatments are still poorly understood. With regard to ICIs in this population, the literature is scarce but nevertheless promising, with case reports suggesting major clinical activity for POLE-mutated CRC patients [52,53,54]. Several ongoing clinical trials are dedicated to this population (NCT03827044, NCT02715284, NCT03012581).
3.2 Tumor Mutational Burden and Efficacy of ICIs in CRC
TMB is an exonic genomic measure of non-synonymous mutations in tumor cells. Using algorithms comparing DNA sequences from control tissues to tumor samples, TMB helps to predict the number of somatic mutations within tumors. Therefore, it is an indirect measure of predicted tumoral neoantigens [55]. The usefulness of TMB is based on the concept that when it is high, the likelihood of observing an effector T cell response directed against a specific tumor antigen is increased. This concept seems to be verified in MSI/dMMR tumors, as both are hypermutated and rich in effector immune cells. A recent study evaluated the predictive value of TMB in MSI/dMMR tumors treated by anti PD-1 [41]. The authors showed that patients with high TMB (> 37 mt/Mb) tumors displayed an ORR of 100% (N = 13) and improved PFS compared with patients with low TMB tumors (N = 9) [39]. While these results are promising, confirmation in prospective larger cohorts is needed before being translated to clinical practice because, as previously mentioned, the impact of the mutation load varies from one study to another.
MSS/pMMR CRC tumors may also display a high TMB but with a low frequency, including exonucleasic domain POLE-mutated tumors and other CRCs in which the mechanism of hypermutation remains unclear. A recent 2:1 randomized phase II clinical trial, CCTG CO.26, assessed the combination of tremelimumab, an anti-CTLA-4 agent, and durvalumab, versus best supportive care (BSC) in 180 MSS chemoresistant mCRC patients [56]. The trial reached its primary endpoint, with a median OS of 6.6 months for the ICI combination versus 4.1 months for BSC (HR 0.72 P = 0.07 for a preplanned threshold < 0.1 for statistical significance) but without any improvement in the median PFS, which was 1.8 and 1.9 months, respectively. DCR was 22.7% for the experimental arm versus 6.6% for BSC. TMB was assessed on cell-free DNA (cfDNA) using a 500-gene next-generation sequencing panel [57], and, excluding two MSI/dMMR patients, the mean TMB was 20.4 ± 16.3 mts/Mb (range: 0.96–114.0). Interestingly, 21% of tumors had a high TMB (defined as ≥ 28 mt/Mb), which was associated with better OS compared to patients with low or intermediate TMB tumors [5.5 months vs. 3 months, respectively; hazard ratio (HR) 0.34, 90% confidence interval (CI) 0.18–0.63]. Nevertheless, the pre-specified threshold of 20 mt/Mb was not predictive of benefit for DCR, PFS, or OS.
Genotyping of circulating tumor DNA (ctDNA) allows for a non-invasive quantification and characterization of the tumoral molecular profile and can indicate tumor heterogeneity and changes in TMB in real time. High blood TMB (bTMB) has been recently associated with an increase of response and survival in other tumor types treated with immunotherapy, becoming a promising biomarker for ICIs [58,59,60]. However, bTMB shows a higher number of mutations per megabase compared to classic TMB assessed in tissue resulting in variable concordance data between tissue and liquid samples. Moreover, in CRC it remains unclear why a benefit is observed in OS but not in other outcomes. Further prospective studies are needed for implementation of bTMB into clinical practice.
The theoretical presence of tumoral neoantigens may not be able to independently predict their immunogenicity. A high number of neoantigens and/or high-quality neoantigens are necessary to induce a durable antitumor immune response. A recent study underlined that non-hypermutated MSS CRCs have a very low predicted number of neoantigens (45 ± 22) compared to MSI (635 ± 308) or hypermutated MSS CRC (1651 ± 1475) [61]. Notably, the number of predicted neoantigens decreases from the early to the advanced stage in the context of an active immunoediting, resulting in the elimination of the immunoreactive subclones and selection of immune privileged CRC subclones [62]. Specific translational immunological studies should be performed at a later stage to better understand the immune escape mechanisms in mCRC.
3.3 Immunoscore® and Efficacy of ICIs in CRC
The Immunoscore® is a measure of the tumor-associated inflammatory stroma, assessing pan-/cytotoxic lymphocyte densities (CD3+ and CD8+, respectively) inside the tumor and at the invasion margin [63]. The Immunoscore® is a validated independent prognostic factor in early-stage CRC for predicting disease-free survival. Patients with a low Immunoscore®, i.e., low infiltration of CD3+ and CD8+ cells, a have higher risk of recurrence, thereby justifying its use as a stratification factor in adjuvant clinical trials [64]. In this publication, when stratified into two Immunoscore® categories (0–1, and 2–4), 49 MSI tumors (16%) had a low Immunoscore® (0–1) and 255 (84%) had an intermediate and high Immunoscore® (2–4). Patients with an intermediate and high Immunoscore® had prolonged DFS and OS, irrespective of their microsatellite status (unadjusted HR for high vs. low Immunoscore® in MSI tumors 0.56, 95% CI 0.34–0.90; P = 0.0150). DFS of patients with a high Immunoscore® was similar for MSS and MSI tumors (around 80% at 3 years) and DFS of patients with a low Immunoscore® was similar for MSS and MSI tumors (around 60%).
The usefulness of the Immunoscore® in advanced disease seems more complex, as it relies on the assessment of the immune contexture of the invasion margin. Indeed, discordances between the primary tumor and the metastases have been observed, and only the Immunoscore® of metastases remained an independent prognostic factor in multivariate analyses [65]. In other tumors, lymphocytic infiltration or intratumoral PD-L1 expression is not always associated with a benefit from immunotherapy, which is better predicted by transcriptional signatures where interferon gamma (IFN-γ) response genes are included [66]. The Immunoscore® correlation with IFN-γ response remains to be established and, if confirmed, would allow the prospective assessment of ICIs according to Immunoscore® in both dMMR and pMMR tumors.
3.4 Molecular Alterations and Immune Escape Mechanisms in CRC
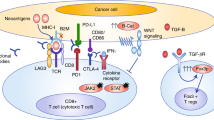
The international consensus molecular classification of CRC has been proposed, with four subtypes described so far: CMS1 (immune), CMS2 (canonical), CMS3 (metabolic), and CMS4 (mesenchymal) [16]. Interestingly, the CMS subtypes are associated with a specific immune and stromal profile [67, 68]. The CMS was developed using a set of localized tumors and its value in the metastatic setting remains controversial: (1) spatial CMS heterogeneity has been described, as some tumors have a homogeneous CMS and others have a heterogeneous CMS, which complicates the allocation of tumors to a specific group and modulates the prognosis [69]; (2) CMS modifications have been observed within metastatic tumors under active antitumoral treatment such as with anti-EGFR agents [70]. Consequently, CMS clinical applicability in the metastatic setting remains limited and even reinforced by the complexity of bioinformatical analyses. Nevertheless, CMS helped to identify at least three main immune escape mechanisms in CRC that are related to distinct molecular pathway alterations. Importantly, there is also an increasing body of evidence showing that the genomic landscape of CRC correlates with specific immune profiles and immune escape mechanisms, which are consistent with the one described in the CMS. Figure 2 proposes an immune classification of CRC based on molecular pathway alterations and the potential treatment strategies to immunomodulate each tumor subtype.

Immune classification of colorectal cancer subtypes based on molecular pathway alteration
Firstly, tumors with DNA repair impairment, such as MMR deficiency or mutations within the polymerases exonucleasic domain, are prone to accumulating immunogenic mutations. These tumors are immune inflamed and characterized by high infiltration in CD8 cells, a high number of neoantigens, and increased expression of CMH-I and immune checkpoints such as PD-1, PD-L1, and CTLA-4, resulting in an immune profile that is highly favorable for ICI activity.
Secondly, CRCs with TGF-β pathway dependency present strong angiogenesis activation and an immune-inflamed profile. The role of TGF-β in colorectal carcinogenesis is complex as it may activate multiple transduction pathways and has a dual anti- and pro-tumoral role from early- to late-stage cancer [71]. Multiple alterations in genes implicated in TGF-β transduction pathways have been described in CRC, such as in the SMAD family genes, BMPR1A or TGFb receptors themselves. The impact of these alterations on immune cell recruitment in human CRCs remains to be elucidated, but multiple preclinical models have shown that impairment of these genes results in a chronic inflammation favoring colorectal cancer progression [71]. Even if immune inflamed, the immune contexture of TGF-β-dependent tumors is different from DNA repair-impaired tumors and is characterized by an imbalance between the pro- and anti-tumoral immune cells with higher Treg, M0 and M2 macrophages, and a lower number of CD8+ cells. Enrichment in immunosuppressive and complement factors such as high expression of chemokines attracting myeloid cells is also observed, reinforcing the pro-tumoral immune response. Recent preclinical studies suggest that targeting the TGF-β pathway in CRC favors the recruitment of T cells and enhances the Th1 response leading to an increased sensitivity to ICI [72]. Another attractive target is angiogenesis, as vascular endothelial growth factor (VEGF) pathway inhibitors have shown encouraging results in other tumor types in combination with ICIs [73, 74]. Indeed, in CRC, anti-VEGF agents were shown to increase CD8 + cells infiltration and decrease Treg in resected metastases, providing a rationale for combination strategies with ICIs [75].
Finally, tumors with WNT/β-catenin pathway activation derived from APC or CTNNB1 mutations have been shown to be poorly infiltrated by immune cells through the lack of immune-cell trafficking [76] and characterized by: (1) a few nonsynonymous mutations; (2) low intratumoral immune infiltrate with a global decrease in lymphocytes, CD8 + cells, Th1, activated NK, and PD-L1 + mono/macrophages; and (3) low CMH-1 and B2M expression, suggesting poor antigen presentation. Preclinical models of CRC suggest that inhibition of the WNT/β-catenin pathway could increase the immunological “hotness” of these tumors and generate sensitivity to ICIs [77].
Compared to CMS assessment, immuno-genomic profiling could be more easily transposable in clinical practice as it relies on DNA sequencing and/or pathological assessment. This immuno-genomic profiling should probably be repeated over time during the course of the metastatic disease and after exposure to treatments due to constant immunoediting, which actively modulates the immuno-genomic phenotype [62].
Setting aside the issue of the CMS classification, a recent early phase I trial assessing the combination of nivolumab and regorafenib (anti-VEGF Receptor inhibitor) in MSS mCRC showed encouraging results with an ORR of 33% and a median PFS of 6.3 months warranting further development [78], suggesting that angiogenesis inhibitors and ICIs are synergistic in a subset of CRC, possibly the TGFβ/angiogenesis subgroup. Nevertheless, the use of other kinase inhibitors, such as MEK inhibitors, in combination with anti PD-L1 failed to show an improvement compared to regorafenib monotherapy in MSS mCRC tumors, despite showing promising results in phase Ib trials [13].
4 Conclusion and Perspectives
ICIs have been a major oncologic breakthrough in dMMR/MSI CRC but showed very limited results in low-TMB pMMR/MSS patients, with disappointing results in several clinical trials. Despite dramatic responses to pembrolizumab or nivolumab in patients with dMMR/MSI tumors, nearly half of the patients will eventually progress, and no biomarker is currently validated to correctly predict the resistance or the benefit derived from ICI, precluding further clinical development. Potential biomarkers are controversial because they are assessed retrospectively or in post hoc translational studies of small cohorts. Two examples are (1) B2M mutations thought to be predictive of resistance in early studies and not confirmed in latter studies, and (2) the discrepancies between mutation load and TMB correlations with clinical outcome, potentially explained by technical limitations and non-consensual bioinformatical analyses. Combination of anti-PD-1/PD-L1 and anti CTLA-4 agents seems to increase the benefit with an acceptable safety profile in selected patients with MSI/dMMR mCRC. Currently, there are still many unresolved questions concerning the clinical management of patients with ICI-treated MSI/dMMR mCRC such as duration of therapy, surgical removal of post-immunotherapy residual lesions, the best predictive factors of efficacy, or the choice of the best treatment scheme approach.
POLE/D exonucleasic domain mutations are an important alternative biomarker for selecting the best candidates for ICIs in MSS/pMMR mCRC. POLE-mutated tumors seem rare and confirming ICI activity in this subset may be challenging prospectively as the pathogenicity of these mutations remains unclear outside of the described hotspots. TMB is an attractive biomarker for offering ICIs to patients with MSI/pMMR CRC with a high mutation load. First results seem to indicate a limited benefit on OS but not on other outcomes. However, TMB assessment remains expensive, without a clear threshold and technical/bioinformatical pitfalls, and its predictive value remains to be established both for MSS/pMMR and MSI/dMMR tumors. The Immunoscore® and IFN-γ response signature are also emerging biomarkers but preclinical/translational studies are still necessary to confirm their value in CRC, especially in pMMR patients, before designing prospective clinical trials. Finally, immuno-genomic classification may be useful, especially to separate MSS/pMMR CRCs in clinical trials in accordance with immune escape mechanisms. Incorporating immune-genomic profiling in future studies assessing immunomodulation/targeted therapeutic combinations would be of added value to identify subsets of patients benefiting from these treatments and move to individualized immunotherapeutic strategies in CRCs.
References
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20.
Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I study of pembrolizumab (MK-3475; Anti–PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015;21:4286–93.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of Anti-PD-L1 Antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30.
Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, et al. Correlation between density of CD8 + T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res. 2015;75:3446–55.
Marisa L, Svrcek M, Collura A, Becht E, Cervera P, Wanherdrick K, et al. The balance between cytotoxic T-cell lymphocytes and immune checkpoint expression in the prognosis of colon tumors. J Natl Cancer Inst. 2018;110:68–77.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51.
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75.
Overman M, Lonardi S, Wong K, Lenz H, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Overman M, McDermott R, Leach J, Lonardi S, Lenz H, Morse M, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20:849–61.
Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6.
Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. J Clin Oncol. 2016;34(suppl):103.
Le DT, Kavan P, Won Kim T, Burge M, Van Cutsem E, Hara H, et al. KEYNOTE-164: Pembrolizumab for patients with advanced microsatellite instability high (MSI-H) colorectal cancer. J Clin Oncol. 2018;36(suppl):3514.
Diaz L, Marabelle A, Kim TW, Geva R, Van Cutsem E, Andre T, et al. Efficacy of pembrolizumab in phase 2 KEYNOTE-164 and KEYNOTE-158 studies of microsatellite instability high cancers. Ann Oncol. 2017;28:386P.
Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability–high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2019. https://doi.org/10.1200/JCO.19.02107.
Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Nivolumab (NIVO) + low-dose ipilimumab (IPI) in previously treated patients (pts) with microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): long-term follow-up. J Clin Oncol. 2019;37(suppl):635.
Overman MJ, Bergamo F, McDermott RS, Aglietta M, Chen F, Gelsomino F, et al. Nivolumab in patients with DNA mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC): long-term survival according to prior line of treatment from CheckMate-142. J Clin Oncol. 2018;36(suppl):554.
Lenz H-JJ, Van Cutsem E, Limon ML, Wong KY, Hendlisz A, Aglietta M, et al. LBA18_PRDurable clinical benefit with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line therapy in microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). Ann Oncol. 2018;29:mdy424.019.
Segal NH, Wainberg ZA, Overman MJ, Ascierto PA, Arkenau H-T, Butler MO, et al. Safety and clinical activity of durvalumab monotherapy in patients with microsatellite instability-high (MSI-H) tumors. J Clin Oncol. 2019;37(suppl):670.
Cohen R, Hain E, Buhard O, Guilloux A, Bardier A, Kaci R, et al. Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of microsatellite instability or mismatch repair deficiency status. JAMA Oncol. 2019;5(4):551–5.
Svrcek M, Lascols O, Cohen R, Collura A, Jonchère V, Fléjou J-F, et al. MSI/MMR-deficient tumor diagnosis: which standard for screening and for diagnosis? Diagnostic modalities for the colon and other sites: Differences between tumors. Bull Cancer (Paris). 2019;106:119–28.
O’Neil BH, Wallmark JM, Lorente D, Elez E, Raimbourg J, Gomez-Roca C, et al. Safety and antitumor activity of the anti–PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS One. 2017;12:e0189848.
Cohen R, Buhard O, Cervera P, Hain E, Dumont S, Bardier A, et al. Clinical and molecular characterisation of hereditary and sporadic metastatic colorectal cancers harbouring microsatellite instability/DNA mismatch repair deficiency. Eur J Cancer. 2017;86:266–74.
Salvador MU, Truelson MRF, Mason C, Souders B, LaDuca H, Dougall B, et al. Comprehensive paired tumor/germline testing for lynch syndrome: bringing resolution to the diagnostic process. J Clin Oncol. 2019;37:647–57.
Kopetz S, André T, Overman MJ, Zagonel V, Lonardi S, Aglietta M, et al. Exploratory analysis of Janus kinase 1 (JAK1) loss-of-function (LoF) mutations in patients with DNA mismatch repair-defıcient/microsat- ellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC) treated with nivolumab plus ipilimumab in CheckMate-142. Proc Am Assoc Cancer Res. 2018;59:2603.
Sveen A, Johannessen B, Tengs T, Danielsen SA, Eilertsen IA, Lind GE, et al. Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 2017;9:46.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–29.
Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188–201.
Middha S, Yaeger R, Shia J, Stadler ZK, King S, Guercio S, et al. Majority of B2M-mutant and -deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis Oncol. 2019. https://doi.org/10.1200/PO.18.00321.
Routy B, Chatelier EL, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Llosa NJ, Luber B, Siegel N, Awan AH, Oke T, Zhu Q, et al. Immunopathologic stratification of colorectal cancer for checkpoint blockade immunotherapy. Cancer Immunol Res. 2019;7(10):1574–9.
Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science. 2019;364:485–91.
Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol. 2019;30:1096–103.
Tachon G, Frouin E, Karayan-Tapon L, Auriault M-L, Godet J, Moulin V, et al. Heterogeneity of mismatch repair defect in colorectal cancer and its implications in clinical practice. Eur J Cancer. 2018;95:112–6.
Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24(9):1449–58.
Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171(1042–1056):e10.
Gerwing M, Herrmann K, Helfen A, Schliemann C, Berdel WE, Eisenblätter M, et al. The beginning of the end for conventional RECIST—novel therapies require novel imaging approaches. Nat Rev Clin Oncol. 2019;16(7):442–58.
Sehdev A, Cramer HM, Ibrahim AAM, Younger AE, O’Neil BH. Pathological complete response with anti-PD-1 therapy in a patient with microsatellite instable high, BRAF mutant metastatic colon cancer: a case report and review of literature. Discov Med. 2016;21:341–7.
Stadler ZK, Battaglin F, Middha S, Hechtman JF, Tran C, Cercek A, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J Clin Oncol. 2016;34:2141–7.
Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7:746–56.
Ahn S-M, Ansari AA, Kim J, Kim D, Chun S-M, Kim J, et al. The somatic POLE P286R mutation defines a unique subclass of colorectal cancer featuring hypermutation, representing a potential genomic biomarker for immunotherapy. Oncotarget. 2016;7:68638–49.
Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16:71–81.
Bourdais R, Rousseau B, Pujals A, Boussion H, Joly C, Guillemin A, et al. Polymerase proofreading domain mutations: new opportunities for immunotherapy in hypermutated colorectal cancer beyond MMR deficiency. Crit Rev Oncol Hematol. 2017;113:242–8.
Domingo E, Freeman-Mills L, Rayner E, Glaire M, Briggs S, Vermeulen L, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1:207–16.
Guerra J, Pinto C, Pinto D, Pinheiro M, Silva R, Peixoto A, et al. POLE somatic mutations in advanced colorectal cancer. Cancer Med. 2017;6:2966–71.
Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Canc Netw. 2017;15:142–7.
Santin AD, Bellone S, Buza N, Choi J, Schwartz PE, Schlessinger J, et al. Regression of chemotherapy-resistant polymerase ε (POLE) ultra-mutated and MSH6 hyper-mutated endometrial tumors with nivolumab. Clin Cancer Res. 2016;22:5682–7.
Bhangoo MS, Boasberg P, Mehta P, Elvin JA, Ali SM, Wu W, et al. Tumor mutational burden guides therapy in a treatment refractory POLE-mutant uterine carcinosarcoma. Oncologist. 2018;23:518–23.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry S, Couture F, et al. CCTG CO.26: Updated analysis and impact of plasma-detected microsatellite stability (MSS) and tumor mutation burden (TMB) in a phase II trial of durvalumab (D) plus tremelimumab (T) and best supportive care (BSC) versus BSC alone in patients (pts) with refractory metastatic colorectal carcinoma (rmCRC). J Clin Oncol. 2019;37(15_suppl):3512. https://doi.org/10.1200/JCO.2019.37.15_suppl.3512.
Artyomenko A, Sikora M, Lefterova M, Raymond VM, Gavino D, Barbacioru C, et al. Microsatellite instability detection by targeted sequencing of cell-free DNA. Ann Oncol. 2018;29:viii400–41.
Wang F, Zhao Q, Wang Y-N, Jin Y, He M-M, Liu Z-X, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019. https://doi.org/10.1001/jamaoncol.2019.2963.
Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24:1441–8.
Khagi Y, Goodman AM, Daniels GA, Patel SP, Sacco AG, Randall JM, et al. Hypermutated circulating tumor DNA: correlation with response to checkpoint inhibitor-based immunotherapy. Clin Cancer Res. 2017;23:5729–36.
Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64.
Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L, et al. Evolution of metastases in space and time under immune selection. Cell. 2018;175(751–765):e16.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4.
Pagès F, Mlecnik B, Marliot F, Bindea G, Ou F-S, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391:2128–39.
Kwak Y, Koh J, Kim D-W, Kang S-B, Kim WH, Lee HS. Immunoscore encompassing CD3 + and CD8 + T cell densities in distant metastasis is a robust prognostic marker for advanced colorectal cancer. Oncotarget. 2016;7:81778–90.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Becht E, de Reyniès A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22:4057–66.
Karpinski P, Rossowska J, Sasiadek MM. Immunological landscape of consensus clusters in colorectal cancer. Oncotarget. 2017;8:105299–311.
Laurent-Puig P, Marisa L, Ayadi M, Blum Y, Balogoun R, Pilati C, et al. 60PDColon cancer molecular subtype intratumoral heterogeneity and its prognostic impact: An extensive molecular analysis of the PETACC-8. Ann Oncol. 2018;29(suppl):mdy269.058.
Woolston A, Khan K, Spain G, Barber LJ, Griffiths B, Gonzalez-Exposito R, et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal cancer. Cancer Cell. 2019;36(35–50):e9.
Jung B, Staudacher JJ, Beauchamp D. Transforming growth factor β superfamily signaling in development of colorectal cancer. Gastroenterology. 2017;152:36–52.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–43.
McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–57.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Manzoni M, Rovati B, Ronzoni M, Loupakis F, Mariucci S, Ricci V, et al. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology. 2010;79:187–96.
Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8:730–49.
Xiao Q, Wu J, Wang W-J, Chen S, Zheng Y, Yu X, et al. DKK2 imparts tumor immunity evasion through β-catenin-independent suppression of cytotoxic immune-cell activation. Nat Med. 2018;24:262–70.
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric (GC) or colorectal cancer (CRC): an open-label, dose-finding, and dose-expansion phase 1b trial (REGONIVO, EPOC1603). J Clin Oncol. 2019;37:2522.
Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–54.
Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15:325–40.
Holubec L, Polivka J, Safanda M, Karas M, Liska V. The role of cetuximab in the induction of anticancer immune response in colorectal cancer treatment. Anticancer Res. 2016;36:4421–6.
Pelster MS, Amaria RN. Combined targeted therapy and immunotherapy in melanoma: a review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther Adv Med Oncol. 2019;11:1758835919830826.
Vanpouille-Box C, Formenti SC, Demaria S. Toward precision radiotherapy for use with immune checkpoint blockers. Clin Cancer Res. 2018;24:259–65.
Weintraub K. Take two: combining immunotherapy with epigenetic drugs to tackle cancer. Nat Med. 2016;22:8–10.
Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11:1–25.
Acknowledgements
We would like to thank Michelle Lamendola-Essel for editing assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this article.
Conflict of Interest
Benoît Rousseau served in a consulting/advisory role for Bayer, Roche, Novartis, Gilead, and Servier and has received travel, accommodation, and other expenses from Bayer, Servier, and Astellas. Joana Vidal has received honoraria for advisory role, speaker or travel grants from Amgen, Hoffman La-Roche, Merck-Serono, Sanofi, and Sysmex Inostics. Romain Cohen has received honoraria from Amgen, Sanofi, and Servier, and travel fees from Sanofi. Thierry André reported receiving honoraria from Roche, Genentech, Ventana, Sanofi, Bristol-Myers Squibb, MSD Oncology, Servier, and Pierre Fabre; consulting or advisory role for Roche, Amgen, Bristol-Myers Squibb, and HalioDX; speakers’ bureau for Bristol-Myers Squibb and Servier; travel, accommodation, and other expenses from Roche, Genentech, Amgen, and Bristol-Myers Squibb. Raphaël Colle and Luis A Jr Diaz declare that they have no conflicts of interest that might be relevant to the contents of this article.
Rights and permissions
About this article

Cite this article
Cohen, R., Rousseau, B., Vidal, J. et al. Immune Checkpoint Inhibition in Colorectal Cancer: Microsatellite Instability and Beyond. Targ Oncol 15, 11–24 (2020). https://doi.org/10.1007/s11523-019-00690-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-019-00690-0