
Abstract
Background
Tasquinimod is a small molecule with immunomodulatory, anti-angiogenic, and anti-metastatic properties that targets the tumor microenvironment. This study aimed to obtain a clinical proof of concept that tasquinimod was active and tolerable in patients with advanced solid tumors.
Patients and Methods
This early stopping design, open-label, proof-of-concept clinical trial evaluated the clinical activity of tasquinimod in four independent cohorts of patients with advanced hepatocellular (n = 53), ovarian (n = 55), renal cell (n = 38), and gastric (n = 21) cancers. Tasquinimod was given orally every day (0.5 mg/day for at least 2 weeks, with dose increase to 1 mg/day) until radiological progression according to Response Evaluation Criteria in Solid Tumor (RECIST) 1.1 criteria, intolerable toxicity, or patient withdrawal. The primary efficacy endpoint was progression-free survival (PFS) rate according to RECIST 1.1 by central assessment.
Results
Interim futility analyses at 8 weeks (6 weeks for the gastric cancer cohort) found adequate clinical activity of tasquinimod only in the hepatocellular cohort and recruitment to the other three cohorts was stopped. PFS rates were 26.9% at 16 weeks, 7.3% at 24 weeks, 13.2% at 16 weeks, and 9.5% at 12 weeks, respectively, in hepatocellular, ovarian, renal cell, and gastric cancer cohorts. The pre-defined PFS threshold was not reached in the hepatocellular cancer cohort at the second stage of the trial. The most common treatment-related adverse events were fatigue (48.5%), nausea (34.1%), decreased appetite (31.7%), and vomiting (24.6%).
Conclusions
This study failed to demonstrate clinical activity of tasquinimod in heavily pre-treated patients with advanced hepatocellular, ovarian, renal cell, and gastric cancer.
Trial registration
NCT01743469.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Tasquinimod is a second-generation oral quinoline-3-carboxamide with multiple effects in the tumor microenvironment that inhibits tumor growth and metastasis [1]. A key target of tasquinimod is S100A9, a multifunctional immunomodulatory protein found in high levels in the microenvironment of several tumor types [1]. S100A9 interacts with proinflammatory receptors, such as receptor for advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4), expressed on myeloid-derived suppressor cells (MDSCs), macrophages, endothelial, and other cells. Tasquinimod binds to S100A9 and inhibits its interaction with RAGE and TLR4 and thereby influences the activation and accumulation of MDSCs in the tumor [2,3,4] Signals produced in the tumor microenvironment appear to stimulate many of the pro-angiogenic functions of the MDSCs [5]. For example, tumor-infiltrating macrophages are stimulated to act as a potent pro-angiogenic force in tumors by exposure to tumor hypoxia and/or such tumor cell-derived cytokines as vascular endothelial growth factor (VEGF), tumor necrosis factor α and angiopoietin 2.
In hepatocellular, ovarian, renal cell and gastric cancers, resistance to treatment and disease relapse or progression is common. Angiogenesis is significantly involved in the development of these four tumor types, and resistance to angiogenesis inhibitors thought to occur by tumor cell adaptation through upregulation of pre-existing redundant or evasive mechanisms [6]. Studies have shown improved progression-free survival (PFS) in patients with metastatic castration-resistant prostate cancer (mCRPC) treated with tasquinimod [7, 8].
This study was undertaken to obtain clinical proof-of-concept that tasquinimod was active and tolerable in patients with advanced hepatocellular (HCC), epithelial ovarian (OC), renal cell carcinoma (RCC), or gastric cancers (GC).
2 Patients and Methods
This phase II, open-label, proof-of-concept clinical trial (ClinicalTrials.gov identifier NCT01743469) was performed at 24 sites in Belgium, Canada, France, Spain, and the UK (supplementary Table S1, available online). The study was conducted under the provisions of the Declaration of Helsinki, and in accordance with the International Conference on Harmonization Consolidated Guideline on Good Clinical Practice. The protocol and amendments and documents for patients were reviewed and approved by an independent ethics committee or institutional review board at each institution prior to study start. Written informed consent was provided by all patients participating in the study,
2.1 Study Participants and Treatment
The study included adult patients with histologically confirmed advanced HCC, OC, RCC, and GC who had progressed during or after standard therapies. Full inclusion and exclusion criteria are provided in supplementary Table S2. All patients were to receive tasquinimod at a starting dose of 0.5 mg/day maintained for at least 2 weeks and then increased to 1 mg/day when possible. The dose could be maintained or reduced in case of Treatment Emergent Adverse Events (TEAEs).
2.2 Study Plan and Design
Tasquinimod treatment was given until radiological progression according to Response Evaluation Criteria in Solid Tumor (RECIST) v1.1 criteria, the criteria validated at the time of the study [9], toxicity, or patient withdraw. Full details of study visit schedule and clinical assessments are provided in supplementary Tables S3 and S4, available online.
This study used a two-stage early stopping design (with a futility analysis at stage 1) to assess the activity of tasquinimod based on the proportion of patients who had neither progressed nor died at pre-defined time points (the PFS rate [10]) in each of the four cohorts. The data applied was based on historical data. For each cohort, the sample size was calculated based on a one-sided α of ≤0.1 and a power of ≥90% together with the constraints that the chance of early stopping given the null hypothesis (i.e. tasquinimod showed inadequate clinical activity) was ≥50% and the chance of early stopping given the alternative hypothesis was ≤10%.
The interim futility analysis was performed for each cohort after a pre-defined number of patients reached Week 8 (Week 6 for the GC cohort; T1). If the number of patients who had not progressed or died at this time was lower than expected so the null hypothesis was not rejected, recruitment was stopped; otherwise, recruitment continued. The expected PFS rate for an active treatment at T1 (and to pursue the recruitment until T2) was 60% for the HCC, 65% for the OC and RCC and 40% for the GC cohort. At T2 the expected PFS rates for an active treatment were 40% for both the HCC and RCC cohorts at 16 weeks, 55% for the ovarian cancer (OC) cohort at 24 weeks and 35% for the gastric cancer (GC) cohort at 12 weeks. The cohort patient numbers, analysis timings and expected PFS rates are shown in supplementary Table S5.
2.3 Efficacy and Safety end Points and Assessments
The primary efficacy end point was the proportion of patients who did not progress nor died (PFS rate) according to RECIST v1.1 by central assessment at the final analysis (T2; see supplementary Table S5 for time points). Secondary efficacy end points were PFS duration, response rate, clinical benefit, time to progression (TTP), and overall survival (OS). A patient was considered to have clinical benefit if a complete response, a partial response or stable disease was observed for ≥12 or 16 weeks (according to each cohort) after the first study medication according to RECIST criteria. All tumor assessments were appraised by investigators and then secondly by a central independent reviewer. Overall response was evaluated using the RECIST v1.1 guideline for all cohorts and also by Choi criteria in HCC cohort.
Safety assessments were performed regularly throughout the study: adverse events (AEs), laboratory test values, ECOG performance status, vital signs, and 12-lead electrocardiographic findings were monitored and recorded. AEs and laboratory tests were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) classification version 4.03 (or severity) and coded using MedDRA dictionary.
2.4 Statistical Analysis
Efficacy and safety analyses were performed on all patients receiving at least one dose of tasquinimod. The primary efficacy analysis was the PFS rate (presented with its 95% confidence interval (CI) calculated using the Clopper-Pearson exact method) according to RECIST v1.1 by central assessment at the final analysis (T2). For each cohort, the primary analysis was performed at T2 by comparing the PFS rate with the pre-specified threshold using a one-sided α of 0.1. To analyze the time-dependent parameters such as PFS, TTP, and OS, the Kaplan-Meier method was used. Safety data were analyzed for the Safety Population, separately for each cohort and in an overall pooled analysis at T2. The safety analysis was based on treatment emergent AEs (TEAEs). For each TEAE, worst NCI CTCAE (Version 4.03) grade per patient was tabulated by System Organ Class (SOC) and Preferred Term (PT). Laboratory values were presented by worst NCI CTCAE grade per patient or descriptive statistics.
3 Results
3.1 Patients and Treatment
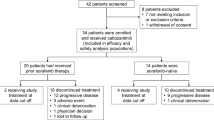
From December 2012 to July 2014, 201 patients were screened, and 167 patients were subsequently enrolled in the four separate cohorts. There were 53 patients with HCC, 55 patients with OC, 38 patients with RCC, and 21 patients with GC. Patient disposition through the study is shown in Fig. 1 and demographic and selected clinical characteristics in the four separate cohorts at baseline are shown in supplementary Table S6.

Patient disposition. ITT, intent to treat; TEAE, treatment-emergent adverse event
Dose escalation to 1 mg/day was achieved in the majority of patients (62–77%), while few required a reduction of treatment dose. The median duration of treatment ranged from 5.9 weeks in the GC cohort to 9.4 weeks in the HCC cohort (supplementary Table S7, available online). Two ovarian and three liver cancer patients were treated with tasquinimod for >1 year.
3.2 Efficacy
For the HCC cohort, the observed PFS rate at T1 was superior to the pre-defined rate required to proceed and more patients were enrolled up to the planned total. For the OC cohort, recruitment as planned was completed before the T1 futility analysis results were available. For the RCC and GC cohorts, the pre-specified PFS rate was not achieved and further recruitment was stopped.
The results presented correspond to the final analysis of all patients enrolled and treated at 12 weeks for the gastric cohort, 16 weeks for renal and hepatic cohorts and 24 weeks for the ovarian cohort. Clinical efficacy parameters are shown in Table 1 for all four cohorts. None of the PFS rates at T2 reached the pre-defined threshold for efficacy.
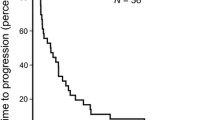
Kaplan-Meier estimates of PFS and OS in the HCC cohort (n = 53) based on central review showed a median (95% CI) of 15.9 (8.0–23.1) and 29.3 (25.0–38.7) weeks, respectively (Fig. 2).

Kaplan-Meier estimates of (a) progression-free survival (PFS) based on central review and (b) overall survival (OS) for the hepatocellular cancer cohort
3.3 Safety
All patients experienced at least one TEAE; serious TEAEs occurred in 26–35% of patients (Table 2). The majority of TEAEs were considered related to study treatment, but most were low grade. Across all patients, the most common TEAEs related to treatment were fatigue (48.5%), nausea (34.1%), decreased appetite (31.7%), and vomiting (24.6%) (Supplemental Table S8, available online).
In all four cohorts, changes in laboratory parameters of grade 3 severity were experienced by less than 5% of patients. Increases in liver function tests were reported in 14 patients (26.9%) in the HCC cohort and occasionally in the other three cohorts. Of particular interest in this study were blood levels of amylase and lipase. Abnormal increases of grade 3 or higher severity were noted in 22 patients for lipase and five patients for amylase (supplementary Table S9, available online).
4 Discussion
The aim of this study was to evaluate tasquinimod activity in patients with solid tumors (liver, ovarian, gastric, and renal cancers). At the time of study design, tasquinimod had only been evaluated in prostate cancer. The study population selected included patients with advanced cancer types with a high unmet medical need reflecting high rates of treatment resistance. In addition, the four advanced cancer disease cohorts were selected because the immunomodulatory, anti-angiogenic, and anti-metastatic properties of tasquinimod underpin a mechanism of action potentially beneficial in these advanced cancers, especially given the lack of standard second- or third-line systemic treatments for these patient populations [1,2,3,4, 8].
The four cohorts can be viewed as separate proof-of-concept studies that were enrolled through a single protocol. While each cohort of patients was analyzed separately for efficacy, the protocol allowed broader capture of information on exploratory end points. Notably, the study incorporated an early stopping design with futility analyses since, with limited data available regarding the efficacy of tasquinimod in other tumor types, exposure to a potentially ineffective drug should be kept to a minimum.
The efficacy analysis revealed that the clinical activity of tasquinimod monotherapy was modest even in the HCC cohort, which proceeded to the second stage of the statistical design based on the T1 analysis. Tasquinimod, with its mechanism of action related to the microenvironment, showed no activity in the selected study population with advanced and resistant disease. In developing drugs of this type, it might be beneficial to evaluate them in patients with minimal residual disease as maintenance therapy after chemotherapy [11].
In previous phase II and III clinical studies, tasquinimod was administered in escalating doses starting with 0.25 mg/day for 2 weeks, followed by 0.5 mg/day for 2 weeks then, with acceptable tolerability, rising to 1.0 mg/day [6, 7]. In the current study, use of 0.5 mg/day as the starting dose allowed dose escalation earlier in the course of treatment and proved to be feasible. The majority of patients then received the higher dose of tasquinimod within a flexible dose regimen in which titration based on individual tolerability mitigated treatment-related toxicities. The overall safety profile of tasquinimod was similar across the four cohorts. As seen in previous studies, tasquinimod has a significant rate of toxicities particularly fatigue, musculoskeletal and gastrointestinal that can result in the need for dose modification [6].
Tasquinimod has been evaluated mainly in metastatic castrate resistant prostate cancer (mCRPC). In chemonaive mCRPC patients, tasquinimod improved radiologic PFS (rPFS) compared with placebo (HR = 0.69, CI 95%: 0.60–0.80), but did not extend OS (HR = 1.10, CI 95%: 0.94–1.28). In a subsequent study in mCRPC patients previously treated with docetaxel, maintenance tasquinimod therapy significantly reduced the risk of rPFS by 40% [12]. In regard to non-solid tumors, tasquinimod has recently received orphan drug status in multiple myeloma by the Food and Drug Administration.
In summary, adequate clinical activity of tasquinimod in patients with advanced HCC, OC, RCC, and GC was not demonstrated in this study. The safety profile of tasquinimod across the four cancer patient cohorts was consistent with that previously reported in mCRPC and no new safety concerns were identified.
References
Raymond E, Dalgleish A, Damber JE, et al. Mechanisms of action of tasquinimod on the tumour microenvironment. Cancer Chemother Pharmacol. 2014;73:1–8.
Källberg E, Vogl T, Liberg D, et al. S100A9 interaction with TLR4 promotes tumor growth. PLoS One. 2012;7:e34207.
Olsson A, Nakhlé J, Sundstedt A, et al. Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J Immunother Cancer. 2015;3:53.
Shen L, Sundstedt A, Ciesielski M, et al. Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine models. Cancer Immunol Res. 2015;3:136–48.
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31.
Bergers G, Hanahan D. Modes of resistance to antiangiogenic therapy. Nat Rev Cancer. 2008;8:592–603.
Pili R, Häggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29:4022–8.
Sternberg C, Armstrong A, Pili R, et al. Phase III, randomized, double-blind, placebo-controlled study of tasquinimod in men with metastatic castrate-resistant prostate cancer. J Clin Oncol. 2016;34:2636–43.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47.
Litwin S, Wong YN, Hudes G. Early stopping designs based on progression-free survival at an early time point in the initial cohort. Stat Med. 2007;26:4400–15.
Khalique S, Hook JM, Ledermann JA. Maintenance therapy in ovarian cancer. Curr Opin Oncol. 2014;26:521–8.
Fizazi K, Ulys A, Sengeløv L et al (2017). A randomized, double blind, placebo controlled phase 2 study of maintenance therapy with tasquinimod in patients with metastatic castration-resistant prostate cancer responsive to or stabilized during first-line docetaxel chemotherapy. Annals Oncol (in press)
Acknowledgements
The authors wish to acknowledge Christelle Descot of Clinical Development Department, Ipsen and Frederique Baton of the Medical Affairs Department, Ipsen, for the contributions to the clinical development program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by Ipsen. Medical writing support provided by Christine McKillop PhD, MedSciMedia Ltd. and funded by Ipsen.
Conflicts of Interest
Bernard Escudier has received consulting fees and honoraria from Novartis, Pfizer, BMS, Exelixis, Bayer, and Roche. Sandrine Faivre has received consulting fees and honoraria from Bayer, Novartis, Merck Serono, Lilly, Ipsen, and Celgene, and support for travel to meetings from Ipsen. Eric Van Cutsem has received grant funding from Ipsen. Nathalie Germann is an employee of Ipsen Innovation and holds stock/share options from Ipsen. Jean-Christophe Pouget is an employee of Ipsen Innovation. Ruth Plummer has received support for travel to meetings and costs relating to recruitment to the study from Ipsen. Fiona Thistlethwaite has received support for travel to meetings and provision of writing assistance from Ipsen. Robert Jones has received grant funding, consulting fees and honoraria, funding for travel, and provision of writing assistance from Ipsen. Julien Edeline has received consulting fees and honoraria from Bayer, BTG, BMS, and Novartis. The other authors declare no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 199 kb)
Rights and permissions
About this article

Cite this article
Escudier, B., Faivre, S., Van Cutsem, E. et al. A Phase II Multicentre, Open-Label, Proof-of-Concept Study of Tasquinimod in Hepatocellular, Ovarian, Renal Cell, and Gastric Cancers. Targ Oncol 12, 655–661 (2017). https://doi.org/10.1007/s11523-017-0525-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-017-0525-2