
Abstract
Aurora kinase inhibitors (AKIs) are a class of antimitotic, small-molecule anticancer agents. MSC1992371A is an AKI being evaluated for the treatment of patients with solid tumors. This phase I, open-label, dose-escalation study determined the maximum tolerated dose (MTD) of MSC1992371A in different dosing schedules in patients with locally advanced or metastatic solid tumors. MSC1992371A was administered on days 1 and 8 (schedule 1) or on days 1, 2, and 3 (schedule 2) of a 21-day cycle. The study was expanded with a third schedule (study drug on days 1–3 and 8–10). Adverse events were monitored throughout the study. Antitumor efficacy, drug pharmacokinetics, and pharmacodynamics were evaluated. Ninety-two patients were enrolled. MSC1992371A was dosed over eight levels in schedules 1 and 2, and the MTD was determined as 74 mg/m2 per cycle for both schedules and as 60 mg/m2 in schedule 3, albeit only in three patients due to discontinuation of the study. Overall, the most common grade 3 or 4 treatment-emergent adverse events were neutropenia, febrile neutropenia, thrombocytopenia, anemia, and fatigue. The most frequent dose-limiting toxicity over all schedules was neutropenia. MSC1992371A plasma concentrations tended to increase with increasing dose levels. Although no complete or partial responses were seen, stable disease ≥3 months was observed in 11 patients. Analysis for markers of target modulation and pharmacodynamics effects was unsuccessful. MSC1992371A was generally well tolerated in patients, with mainly transient hematologic toxicities apparent at an MTD of 60–74 mg/m2/21-day cycle, independent of dosing frequency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitosis, the final phase of the cell division cycle, has been successfully targeted by cancer drugs such as those derived from the vinca alkaloids and the taxanes. These agents disrupt intracellular microtubule dynamics, activate the mitotic checkpoint, block cell division after DNA duplication, and lead to cell death [1]. However, disrupting microtubules is associated with significant neurotoxicity, myelosuppression, mucositis, and neutropenia. Therefore, other mitotic targets have been pursued. The mammalian mitotic kinases, aurora kinases A, B, and C, have been identified as important regulators of mitosis. They are involved in several different steps such as centrosome duplication, formation of a bipolar mitotic spindle and chromosome alignment on the spindle, and in the surveillance of the mitotic checkpoint [2–4]. These serine/threonine kinases are overexpressed in many types of solid tumors including those of the breast, ovary, colon, and liver, and they have been suggested as appropriate targets for small-molecule anticancer drugs [5]. MSC1992371AFootnote 1 (formerly known as AS703569) is a potent, adenosine triphosphate-competitive inhibitor of mammalian aurora kinases A, B, and C as well as several other kinases involved in cell proliferation, including AMP-activated protein kinase, fibroblast growth factor receptor 3, and Janus kinase 2.
MSC1992371A is orally available and has demonstrated significant activity, both as a single agent and in combination with several commonly used chemotherapeutic agents, against a range of solid and hematologic tumors in preclinical models [6]. In these models, activity and toxicity appeared to be schedule dependent with an indication that repeated daily dosing had a better therapeutic window over single high doses. The antiproliferative effects observed were due to disruption of mitosis, which results in irreversible DNA endoreduplication and ultimately apoptosis [6].
Here, we present the results of the first phase I study of MSC1992371A, carried out to determine the maximum tolerated dose (MTD) in different dosing schedules in patients with locally advanced or metastatic solid tumors.
Patients and methods
Patients
The study included patients with histopathologically confirmed locally advanced or metastatic solid tumors that were refractory to standard treatment or for whom therapeutic options were not available. All eligible patients were over 18 years of age, had an Eastern Cooperative Oncology Group performance status of 0–2, and gave written informed consent. Using criteria that are standard in phase I studies, patients were excluded if there was evidence of impaired bone marrow, renal or hepatic function, coagulation disorder, or history of uncontrolled central nervous system metastasis. Patients who had received chemotherapy or investigational anticancer agents within 28 days of study onset were excluded, as were those with prior radiation therapy involving more than 30 % of bone marrow reserves and those who had previously undergone a marrow or stem cell transplant.
Study objectives
The primary aim of the present phase I, first-in-human study was to determine the MTD of MSC1992371A in different dose schedules in patients with advanced solid tumors. Secondary objectives were to provide a preliminary evaluation of the safety, pharmacokinetic (PK), pharmacodynamic (PD), and antitumor effects of MSC1992371A in these patients. The study was conducted at two sites in the USA.
Study design
This was a phase I, open-label, three-schedule, dose-escalation study in patients with solid tumors. Cohorts of three patients with advanced or metastatic solid tumors were sequentially assigned to one of two dose schedules (schedules 1 and 2) and received monotherapy with MSC1992371A in escalating doses. MSC1992371A was administered orally on days 1 and 8 (schedule 1) and on days 1, 2, and 3 (schedule 2) of a 21-day cycle (Fig. 1). The total dose by cycle was the same for both schedules. With schedule 1, the starting dose level based on preclinical toxicology findings was 3 mg/m2/day; with schedule 2, it was 2 mg/m2/day.

Study design
For each dose schedule, dose escalation followed a modified Fibonacci scheme with increments of 100 % (from dose 1 to dose 2), 65 % (from dose 2 to dose 3), 52 % (from dose 3 to dose 4), and 40 % (from dose 4 to dose 5), with subsequent doses increased by 33 %. For schedule 3, the dose was increased by 40 % from dose 1 to dose 2 and then by 33 % for subsequent cycles. A sequential 3 + 3 design was used to define the MTD for each dose schedule according to the number of dose-limiting toxicities (DLT) observed. DLTs were evaluated during the first treatment cycle only, and no dose modifications were allowed during this cycle. If no patient experienced a DLT over 21 days at the lowest dose, a new cohort of three patients was enrolled at the next dose level. If one of three patients experienced a DLT, a further three patients were enrolled at that dose. Escalation proceeded only if there were no further DLTs. In the case of two or more DLTs at a dose level, the dose of MSC1992371A was reduced to the next lower dose level. This dose was expanded and declared the MTD. Patients were treated until disease progression or development of unacceptable toxicity.
After enrollment into the first two schedules was complete, a third dose schedule (with MSC1992371A given on days 1, 2, and 3 and 8, 9, and 10 of a 21-day cycle) was investigated. This third schedule, involving more frequent dosing, was added since few signals of antitumor activity had been seen with schedules 1 and 2. Preclinical data, and experience from the phase I study in hematologic malignancies that was ongoing at the time, suggested that more frequent dosing of MSC1992371A might enhance antitumor activity. Patients accrued to schedule 3 were started at a dose of 10 mg/m2/day, giving a total dose in the first cycle of 60 mg/m2/day. Following a review of the clinical and PD data available from all ongoing clinical studies, the sponsor, in agreement with the investigators, decided to halt recruitment before the MTD with this schedule had been determined.
The study was conducted in accordance with the Declaration of Helsinki, the International Conference for Harmonization Harmonized Tripartite Guideline for Good Clinical Practice, and all applicable regulatory requirements. Approval of the protocol, protocol amendments, and procedures for obtaining informed, written patient consent was given by the institutional review boards of participating institutions.
Safety and definition of DLT and MTD
The safety of MSC1992371A was evaluated throughout the study by a safety monitoring committee. Safety monitoring involved the evaluation of DLTs, the type and number of treatment-emergent adverse events (TEAEs), serious adverse events, physical examinations (including 12-lead electrocardiograms and echocardiograms or multigated angiocardiography), and regular monitoring of clinical laboratory parameters in blood and urine. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 9.1 and graded according to the Common Terminology Criteria for Adverse Events version 3.0.
DLTs were defined as any of the following: grade 4 neutropenia lasting more than 5 days; grade 3 or 4 neutropenia with fever and/or infection; grade 4 thrombocytopenia (or grade 3 thrombocytopenia with bleeding); any grade 3 or 4 nonhematologic toxicity (except grade 3 increase in transaminase lasting less than a week or suboptimally treated grade 3 nausea and vomiting); any grade 2 neurologic toxicity; and any delay in treatment lasting more than 2 weeks due to MSC1992371A-related toxicity.
If more than one of three or one of six patients experienced a DLT at a given dose, up to 12 additional patients were enrolled at the previous dose level, which was considered the MTD. The dose-escalation analysis set, from which the MTD was determined, consisted of all patients who experienced any DLT during cycle 1 and patients who received ≥90 % of all planned study drug doses during cycle 1.
PK parameters and the effect of food
Plasma and urine samples were collected and regularly reviewed throughout the study to determine single-dose and multiple-dose PK parameters for MSC1992371A. Key PK parameters assessed included maximum serum concentration (C max), time to reach C max (t max), area under the serum concentration–time curve from time zero to the last sampling time (AUC0–t ), and apparent terminal half-life. The lower limit of quantification in plasma was 0.05 ng/mL. Plasma samples were collected at the nine time points shown in Fig. 2. Urine was collected pre-dose and over 0–4 and 4–24 h postdose on day 1 of cycles 1 and 2. PK parameters were calculated using Kinetica 4.4.1 software.

Mean plasma concentrations of MSC1992371A according to dose level on day 1 of cycle 1 in fasting patients treated according to schedules 1–3
After determination of the MTD in schedules 1 and 2, the effect of food on MSC1992371A pharmacokinetics was evaluated in the six to nine patients recruited to the expanded MTD cohort. Patients were randomly assigned using a 1:1 ratio to receive either a high-fat, high-calorie breakfast or to fast before dosing. Using a crossover design, patients who fasted in cycle 1 received breakfast before dosing in cycle 2 and vice versa. For cycles beyond cycle 2, all patients remained fasted.
Antitumor activity
The antitumor activity of MSC1992371A was assessed from computed tomography and magnetic resonance imaging scans using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 [7]. Tumor response was evaluated every second cycle. Duration of response was calculated for patients who experienced a complete or partial response (CR, PR).
PD parameters
A series of PD parameters was measured before and during the course of treatment in order to explore for markers expected to be associated with pharmacodynamics effects, antitumor activity, or potential predictors of toxicity. Changes in PD markers were assessed in circulating tumor cells (CTCs). Markers of cell death (cytokeratin 18 [M65] and cytokeratin 18 cleavage [M30]) were measured in plasma. Histone 3 phosphorylation (pHH3) was investigated by immunohistochemistry (IHC) in skin biopsies. The copy number of the aurora kinase A gene was assessed by fluorescence in situ hybridization (FISH). Mutations in p53 were also investigated.
Statistics methods
Continuous variables were summarized using mean, standard deviation, median, minimum, and maximum values. Continuous variables were categorized when applicable into grouped intervals for analysis, with frequencies and percentages. Categorical variables were tabulated using frequencies and percentages. All patients who received at least one dose of study drug and had at least one follow-up visit were included in the safety analysis. The efficacy analysis included all patients who received at least one dose of drug and had at least one objective assessment of response according to RECIST. All analyses were undertaken using SAS version 9.2.
Results
Patient characteristics
A total of 92 patients were treated in the study: 42 patients in schedule 1, 39 in schedule 2, and 11 in schedule 3. The dose-escalation analysis set included 83 patients in total; 38, 38, and 7 in schedules 1, 2, and 3, respectively. Schedules 1 and 2 were escalated through eight dose levels rising from 6 mg/m2 to approximately 100 mg/m2 per 21-day cycle. Schedule 3 was dosed at 60 and 83 mg/m2 over a 21-day cycle before discontinuation of the study.
Baseline characteristics for the patients enrolled in each of the three schedules were comparable (Table 1). Across all three schedules, the majority of patients were Caucasian (92 %) and female (57 %). Patients’ median age was 63 years. The most common types of solid tumors were colorectal (24 % of patients), prostate and breast (14 % each), and lung (9 %). Patients were a median of 47 months from tumor diagnosis in schedules 1 and 2 and 30 months in schedule 3. All patients were heavily pretreated, having received a median of 3 (range 1–11) prior lines of anticancer therapy (excluding antihormonal therapy).
DLTs and MTD
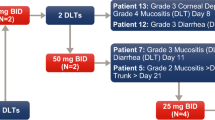
The number of patients treated at each dose level for each of the three schedules and the number of DLTs observed are given in Table 2. With schedule 1, no DLTs occurred until the dose of 49.3 mg/m2/day, at which point two of six patients experienced a DLT of grade 4 neutropenia, considered probably related to study drug. Additional patients were therefore enrolled at the previous dose level but none had a DLT. The dose of 37.1 mg/m2 given on days 1 and 8 (i.e., 74 mg/m2/cycle) was therefore defined per protocol as the MTD.
With schedule 2 (i.e., drug administered on days 1, 2, and 3), no DLTs were experienced until the daily dosage reached 32.9 mg/m2, at which point, three of six patients experienced a DLT (two patients with grade 3 febrile neutropenia and one patient with grade 3 and 4 thrombocytopenia). Increased aspartate aminotransferase and blood alkaline phosphatase levels in one patient in schedule 2 were initially thought to be DLTs, but these hepatic enzyme elevations were retrospectively considered to be unrelated to study drug and due to disease progression. None of the additional patients recruited at the previous dose level of 24.7 mg/m2 had a DLT. This level—at which total dosage per cycle was again 74 mg/m2—was therefore also defined as the MTD.
In schedule 3, in which drug was given on days 1, 2, 3, 8, 9, and 10, no DLTs were observed in the three evaluable patients treated at 10 mg/m2/day (60 mg/m2/cycle). Two of five evaluable patients experienced a DLT (one patient with grade 4 neutropenia and one patient with grade 3 nausea and vomiting) after escalation to 14 mg/m2/day (84 mg/m2/cycle), formally fulfilling for this dose level the criteria for a dose level above the MTD. Although the 10-mg/m2/day dosing seems to be tolerable on this schedule, due to the discontinuation of the study, no additional patients were recruited at this dose level, and therefore, the MTD was not formally confirmed for this schedule.
Safety evaluation across cycles
Overall, 109 cycles of schedule 1 and 122 cycles of schedule 2 were completed. Two cycles of treatment were completed by 55 % of schedule 1 patients and 49 % of patients on schedule 2. The median number of cycles completed per patient was 3 for both schedules (range 0–15 in schedule 1 and 1–22 in schedule 2). For schedule 3, the median number of cycles was 1 (range 0–3). Overall, compliance levels were high with all three treatment schedules. In cycle 1, on which MTDs were based, total median compliance was >98 % for schedules 1 and 2 and 90 % for schedule 3.
All patients in each of the three treatment schedules experienced at least one TEAE. Hematologic TEAEs of grade 3 or 4 are shown in Table 3. Neutropenia grades 3–4 occurred in 19 and 18 % of schedule 1 and 2 patients, respectively. However, febrile neutropenia (2 % in schedule 1, 5 % in schedule 2) was uncommon in the absence of prophylactic myeloid colony-stimulating factors, as was grade 3–4 anemia or thrombocytopenia.
Table 4 shows the most frequently reported nonhematologic TEAEs (all grades). The most common TEAEs were gastrointestinal (GI): nausea and vomiting occurred in 23–59 % of treated patients, and diarrhea, abdominal pain, and constipation were also evident. The most common non-GI TEAE was fatigue, which was experienced by 24 and 44 % of patients treated with schedules 1 and 2, respectively. GI toxicity (diarrhea, nausea, anorexia) and fatigue appeared to be higher in schedule 2 compared to schedule 1, and in schedule 3, although the number of patients was smaller.
Overall, approximately 43 % of patients in schedules 1 and 2 and 64 % of patients in schedule 3 experienced at least one TEAE of grade ≥3. The most frequent grade 3 or 4 events were hematologic, mainly neutropenia with 12/92 (13 %) patients overall experiencing grade 4 neutropenia. Individually, severe nonhematologic toxicities were infrequent and in most cases not related to study drug: in schedule 1 (n = 42), there were two cases of grade 3 vomiting (5 % of patients) and one of grade 4 pulmonary embolism (3 %). Two patients had grade 3 elevations of hepatic enzymes and two had elevated bilirubin (the latter being related to study drug). With schedule 2 (n = 39), there were three cases of drug-related grade 3 fatigue (8 %), two of grade 3 asthenia, and two of grade 4 elevation of aspartate aminotransferase. Grade 3 drug-related fatigue occurred in 18 % of schedule 3 patients, but the number of cases (2 of 11 evaluable patients) was small. Within each schedule, TEAE grade ≥3 occurred more frequently at the higher dose levels of each schedule. In schedule 1, all nine grade 4 events occurred at 74.2 or 100 mg/m2/cycle. In schedule 2, 13/17 (76 %) of TEAE grade ≥3 occurred in dose levels 74.1 and 99 mg/m2/cycle, and in schedule 3, 4/7 (57 %) of TEAE grade ≥3 occurred at the highest dose level (84 mg/m2/cycle).
TEAEs leading to treatment discontinuation occurred in a total of six patients (four in schedule 1 and one each in schedules 2 and 3). These TEAEs were considered drug related in only two cases: thrombocytopenia at a dose of 99 mg/m2 in schedule 2 and nausea and vomiting at a dose of 84 mg/m2 in schedule 3. The six deaths within the reporting period were all associated with disease progression rather than the study drug.
PK profile and the effect of food
PK samples were obtained up to 24 h after administration of MSC1992371A. Moderate to high interindividual variations were observed in the rate of absorption of MSC1992371A (C max and t max) and the extent of drug exposure (area under the plasma concentration curve from administration to last observed concentration at t (AUC0–t ). Maximum plasma concentrations were reached between 1.5 and 4 h (range 0.5–8 h) after first oral administration and were comparable across schedules (Fig. 2a–c). Mean plasma concentrations of MSC1992371A rose with dose level, and therefore, a (non-dose proportional) trend toward increasing exposure was observed with increasing dose.
In all schedules, the extrapolated part of the exposure (AUCextra) exceeded 20 % of the total exposure (AUC0 − ∞) in the majority of the patients, with AUCextra values up to 82 %. Due to these limitations, the terminal elimination phase of MSC1992371A could not be estimated precisely, and therefore, t 1/2 and AUC0 − ∞, apparent clearance and volume of distribution could not be reliably determined in this trial. However, the main PK findings for MSC1992371A in schedules 1 and 2, summarized in Table 5, show the effect of dosing at the MTD in the fed and fasted state. The results of the substudy suggested that food taken before dosing delayed and lowered peak plasma drug concentrations when compared to the fasted state and potentially led to a lower AUC.
The cumulative amount of MSC1992371A excreted in urine over 24 h accounted for less than 3 % of the administered dose in all individuals. Renal excretion is therefore only a minor pathway of elimination for this drug.
Antitumor activity
The best overall response to treatment among the 73 evaluable patients was stable disease in 14 of 38 patients (37 %) in schedule 1, 15 of 31 patients (48 %) in schedule 2, and 1 of 4 patients (25 %) in schedule 3. Four patients (11 %) in schedule 1 and seven patients (23 %) in schedule 2 showed stable disease for at least 3 months. In schedule 3, all four evaluable patients progressed within the first three cycles of treatment. No patient on any of the schedules or doses evaluated in this trial had a confirmed CR or PR.
Although signs of antitumor activity were limited, two patients showed significant tumor shrinkage. In schedule 1, significant tumor shrinkage (−23 % by RECIST) was observed in a heavily pretreated patient with metastatic breast cancer, initially treated with tamoxifen and chemotherapy, who had then received, for advanced disease, four lines of chemotherapy regimens and two investigational agents. In the course of this study, the patient was treated with MSC1992371A 27.9 mg/m2/day (55.8 mg/m2/21-day cycle) for 4 months (six cycles) until disease progression.
In addition, a patient in schedule 2 with a neuroendocrine tumor treated at the MSC1992371 MTD (i.e., 24.7 mg/m2/day; 74.1/mg/m2/21-day cycle) remained on study for 18 months with a minor response (−17 % by RECIST). This patient had received two prior lines of chemotherapy and one investigational agent with stable disease as best response. The patient discontinued MSC1992371 treatment for progressive disease after 22 cycles.
PD
Biomarker results did not provide evidence to support a relationship between MSC1992371A administration and levels of CTCs. In addition, the measured levels of M30 and M65 did not reveal any association between MSC1992371A and cell death. IHC and FISH assays performed on skin and tumor biopsies did not show MSC1992371A-related modulation of the measured biomarkers (pHH3, AURKA, AURKB, and p53). Because few signs of clinical activity were observed in this study, the possible role of biomarkers as predictors of efficacy and response was not investigated further.
Discussion
Given the role of aurora kinases in regulating mitosis, there was a clear rationale for investigating their inhibitors as anticancer agents [5]. The aurora kinase inhibitor MSC1992371A has shown potential in preclinical models [6]. This phase I study evaluated the MTD of the drug given according to different dosing schedules in patients with locally advanced or metastatic solid tumors that were refractory to or ineligible for standard treatments.
The rationale for testing different dosing schedules of MSC1992371A was based on preclinical studies showing that toxicity depended on both the dose and the frequency of drug administration. Toxicity was typically delayed in onset, reversible, and to a great extent modifiable by dose scheduling. This study was therefore initially designed to assess two different, clinically relevant schedules that provided equivalent total doses of study drug over a 21-day treatment cycle. A third schedule was added to the study to determine whether more frequent dosing of MSC1992371A could enhance the antitumor effect. Sequential dosing (on the first 3 days per 21-day cycle) and intermittent dosing (with drug given on days 1 and 8) had the same MTD and a similar toxicity profile. The third schedule was not fully evaluated but appeared to have a similar safety profile to the other two schedules.
The majority of grade 3 or 4 TEAEs were hematologic and occurred at higher doses. The main DLT for the three schedules was grade 4 neutropenia or febrile neutropenia. Most of the nonhematologic TEAEs were gastrointestinal (nausea, vomiting, abdominal pain), although cases of drug-related fatigue, as well as asthenia and grade 3 liver enzyme elevations, were reported.
Subsequent to this study, there have been clinical studies of MSC1992371A in combination with gemcitabine in patients with advanced solid tumors [8, 9] and as monotherapy in patients with hematologic malignancies such as acute myeloid leukemia and myelodysplastic syndromes [10].
In addition, several other aurora kinase inhibitors (AKIs) are currently in clinical development for the treatment of advanced solid tumors such as colorectal or pancreatic cancer [11–16]. These AKIs have different specificities for the aurora kinase subtypes. Some, such as AZD1152, are specific inhibitors of aurora kinase B [11, 13]; others, such as MLN8054, are specific for aurora kinase A [14]; and others still—such as MSC1992371A—are pan-aurora kinase inhibitors [16]. The reported toxicity of agents targeting aurora kinase appears to vary according to their specificity and may in some cases be overshadowed by off-target effects such as benzodiazepine-like adverse effects with somnolence for one compound [14, 15]. Inhibition of the aurora kinase B subtype is consistently associated with hematologic toxicities such as neutropenia and GI adverse events [13]. The pan-aurora kinase inhibitor investigated in this study, MSC1992371A, has a toxicity profile similar to that reported for the other pan-aurora kinase inhibitors MK0457 [16] and danusertib (PHA739358) [17, 18]. The most common adverse events result from aurora kinase B inhibition are neutropenia, vomiting, nausea, and fatigue, with DLTs of neutropenia or febrile neutropenia reported in phase I trials in patients with solid tumors [16, 17].
Phase I studies in patients with solid tumors treated with one of the above-mentioned AKIs all show modest signs of antitumor activity with only occurrences of prolonged stable disease and inconsistent PD effects [12, 13, 15–17, 19]. These results are in line with the antitumor activity and the PD effects seen for MSC1992371A in this study.
In conclusion, oral MSC1992371A has mainly transient and manageable hematologic and—to a lesser extent—GI dose-limiting toxicity becoming evident at higher doses and with more intense dosing regimens in patients with solid tumors refractory to standard cancer treatments. No objective responses were seen at doses up to and including the MTD. The evaluation of a schedule involving more frequent administration was incomplete, but sufficient to conclude that the MTD was similar and that significant differences in clinical activity were unlikely to be observed. The study of the effect of food on PK showed that in the fasted state, absorption was faster and higher.
In addition to this study, the use of MSC1992371A has been investigated in patients with solid tumors in combination with gemcitabine and in patients with hematologic malignancies as monotherapy over prolonged periods of time [9, 10].
Antitumor activity of MSC1992371A dosed at the MTD appeared modest. Moreover, PD markers have been extensively investigated and have failed to provide guidance for dosing, scheduling, or patient/tumor type selection in any of the conditions studied. The clinical development of this agent has currently been halted. Therefore, additional research will be needed to provide a better insight in the clinical activity of MSC1992371A and other aurora kinase inhibitors for the treatment of advanced malignancies.
Notes
The compound is also known as R763. Rights to this compound are currently owned by Rigel Pharmaceuticals Inc., South San Francisco, CA, USA.
References
Yue QX, Liu X, Guo DA (2010) Microtubule-binding natural products for cancer therapy. Planta Med 76:1037–1043
Adams RR, Carmena M, Earnshaw WC (2001) Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol 11:49–54
Carmena M, Earnshaw WC (2003) The cellular geography of aurora kinases. Nature Rev Mol Cell Biol 4:842–854
Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2:21–32
Keen N, Taylor S (2004) Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 4:927–936
McLaughlin J, Markovtsov V, Li H et al (2010) Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. J Cancer Res Clin Oncol 136:99–113
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Awada A, Alexandre J, Gianella-Borradori A et al. (2010) Phase I and pharmacokinetic (PK) study of two regimens combining the aurora kinase inhibitor AS703569 and gemcitabine in patients with advanced solid tumors. Presented at the 101st Annual Meeting of the American Association of Cancer Research; April 17–21, 2010; Washington, DC [Abstr 2754]
Raymond E, Alexandre J, Besse-Hammer T et al (2013) A phase I and schedule dependency study of the aurora kinase inhibitor MSC1992371A in combination with gemcitabine in solid tumors. Invest New Drugs. doi:10.1007/s10637-013-9950-y
Sonet A, Graux C, Maertens J et al. (2008) Phase l, dose-escalation study of 2 dosing regimens of AS703569, an inhibitor of aurora and other kinases, administered orally in patients with advanced hematological malignancies. Blood 112 [Abstr 2963]
Azzariti A, Bocci G, Porcelli L et al (2011) Aurora B kinase inhibitor AZD1152: determinants of action and ability to enhance chemotherapeutics effectiveness in pancreatic and colon cancer. Br J Cancer 104:769–780
Diamond JR, Bastos BR, Hansen RJ et al (2011) Phase l safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and Aurora kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 17:849–860
Boss DS, Witteveen PO, van der Sar J et al (2011) Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Ann Oncol 22:431–437
Macarulla T, Cervantes A, Elez E et al (2010) Phase l study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol Cancer Ther 9:2844–2852
Dees EC, Infante JR, Cohen RB et al (2011) Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol 67:945–954
Traynor AM, Hewitt M, Liu G et al (2011) Phase l dose escalation study of MK-0457, a novel Aurora kinase inhibitor, in adult patients with advanced solid tumors. Cancer Chemother Pharmacol 67:305–314
Cohen RB, Jones SF, Aggarwal C et al (2009) A phase l dose-escalation study of danusertib (PHA-739358) administered as a 24-hourinfusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clin Cancer Res 15:6694–6701
Steeghs N, Eskens FA, Gelderblom H et al (2009) Phase l pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol 27:5094–5101
Schwartz GK, Carvajal RD, Midgley R et al (2013) Phase I study of barasertib (AZD1152), a selective inhibitor of Aurora B kinase, in patients with advanced solid tumors. Invest New Drugs 31:370–380
Acknowledgments
The trial was sponsored by EMD Serono Inc., Rockland, MA, USA. The authors would like to thank the patients and their families without whom this study would not have been possible. Kellie Hazell and Pat Shannon, RN, from Pinnacle Oncology Hematology, Scottsdale, AZ, USA, have provided administrative assistance with data management. Editorial assistance in the preparation of this manuscript was provided by Margot Eggermont, PhD, and Rob Stepney, both of TRM Oncology, The Hague, The Netherlands, and funded by EMD Serono Inc., Rockland, MA, USA.
Conflict of interest
A. Mita, J. Sarantopoulos, and K. Sankhala have no relevant financial relationships to disclose. M. Mita, M. Gordon, and D. Mendelson have received a study grant from Merck Serono S.A., Geneva, Switzerland. N. Rejeb is and A. Gianella-Borradori and V. Jego were employees at Merck Serono S.A., Geneva, Switzerland. M. Gordon and D. Mendelson have performed consultancy work outside the submitted work for Merck KGaA, Darmstadt, Germany, and Merck Serono S.A., Geneva, Switzerland, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mita, M., Gordon, M., Rejeb, N. et al. A phase l study of three different dosing schedules of the oral aurora kinase inhibitor MSC1992371A in patients with solid tumors. Targ Oncol 9, 215–224 (2014). https://doi.org/10.1007/s11523-013-0288-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-013-0288-3