
Abstract
This study was conducted to describe the modulation of plasma epidermal growth factor receptor (EGFR) ligands in EGFR-positive metastatic colorectal cancer (mCRC) patients during treatment with cetuximab and irinotecan and to explore the clinical implication of plasma levels’ variations as potential biomarkers of benefit. Plasma amphiregulin (AR), epidermal growth factor (EGF), transforming growth factor-α, and heparin binding-EGF were assessed by ELISA in 45 chemorefractory mCRC patients, treated with cetuximab and irinotecan. Plasma levels were measured before and 1 h after the first administration of cetuximab, before and 1 h after the second administration, and before the third and the fifth cycles. KRAS and BRAF mutational status were determined. EGFR ligands’ levels were differently modulated according to tumor KRAS and BRAF mutational status. In KRAS wild-type patients (n = 34), AR and EGF early increased and higher increases were significantly associated with worse clinical outcome. By adopting a specific cut-off value, patients with higher levels of AR 1 h after the first administration had significantly worse response rate, progression free survival, and overall survival. This hypothesis-generating study shows that EGFR ligands are significantly modulated by cetuximab plus irinotecan according to KRAS and BRAF mutational status, and they warrant further investigation as pharmacodynamic markers of resistance to anti-EGFRs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The inhibition of epidermal growth factor receptor (EGFR) by means of monoclonal antibodies (MoAbs) binding the receptor’s extracellular domain has proven to be a potentially efficacious strategy in the treatment of KRAS wild-type metastatic colorectal cancer (mCRC) patients [1–4]. In an effort to further disclose the mechanisms of intrinsic and acquired resistance to anti-EGFR MoAbs, the comprehensive investigation of the “EGFR axis” may provide novel insights. Indeed, while a relevant amount of experiences have focused on the potential role of downstream intracellular signaling pathways of EGFR [5, 6], the contribution of EGFR endogenous ligands has been investigated to a much smaller extent.
Khambata-Ford and colleagues [7] firstly reported that amphiregulin (AR) and epiregulin (ER) tumor mRNA expression levels might represent promising predictors of benefit from anti-EGFR MoAbs, by reporting the association of higher levels of tissue mRNA with better disease control rate and progression free survival (PFS) in a retrospective cohort of chemorefractory mCRC patients treated with cetuximab monotherapy [7]. These results were also confirmed in patients with KRAS wild-type disease [8]. A significant relationship between tissue levels of AR and ER mRNA and clinical outcome was also reported in a series of 121 irinotecan-refractory KRAS wild-type patients, treated with the combination of cetuximab and irinotecan [9]. Moreover, preliminary pharmacoproteomic analysis from a subset of 45 patients enrolled in a phase I dose escalation trial of cetuximab monotherapy described a significant upregulation of plasma circulating AR, transforming growth factor-α (TGF-α), and epidermal growth factor (EGF) levels and found that the increase in plasma concentrations of AR after 6 weeks of treatment was significantly weaker in patients responding to cetuximab monotherapy [10].
The present experience describes the modulation of plasma EGFR ligands occurring during the treatment with cetuximab and irinotecan in a cohort of pretreated, irinotecan-refractory mCRC patients [11]. The aims of this study were to investigate possible differences associated to tumor genetic characteristics (i.e., KRAS and BRAF mutational status) and to explore the potential clinical implication of plasma levels’ variations as pharmacodynamic markers of intrinsic and acquired resistance to anti-EGFRs.
Patients and methods
Study population and design
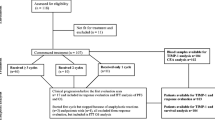
This was an exploratory and hypothesis-generating study with a prospectively planned plasma samples collection. The number of patients (n = 45) to be included was originally defined taking into account the accrual capacity of the single participating institution. As the study was exploratory in nature, no formal statistical hypothesis was postulated. However, the enrolled number of patients is consistent with the entropy-based approach to sample size in translational clinical trials as proposed by Piantadosi [12].
The main patients’ inclusion criteria were the following: histologically confirmed EGFR-positive adenocarcinoma and availability of tissue for molecular analyses, irinotecan-refractory mCRC (whose disease had progressed during or within 3 months after treatment with an irinotecan-based regimen), and measurable disease. Treatment consisted of biweekly cetuximab and irinotecan (cetuximab 500 mg/m2 IV, day 1; irinotecan 180 mg/m2 IV, day 1 every 2 weeks). Tumor response was evaluated every 8 weeks by CT scan according to RECIST 1.0. Investigators performing molecular analyses were blind to clinical data. The ethics committee of the University of Pisa approved the protocol, and the patients provided written informed consent to plasma sampling and genetic analyses (Eudract 2008-003160-19).
Blood samples collection and plasma AR, EGF, TGF-α, and heparin binding-EGF detection
Venous blood was drawn at the following time points:
-
At baseline, before the first administration (d1)
-
One hour after the end of the first infusion of cetuximab (d1-1hr)
-
Before the second administration at day 15 (d15)
-
One hour after the end of the second infusion of cetuximab at day 15 (d15-1hr)
-
Before the third administration at day 29 (d29)
-
Before the fifth administration at day 57 (d57)
Blood samples were immediately centrifuged at 4 °C and plasma fractions were divided in five equal aliquots, frozen, and stored at −80 °C until assayed. Each plasma sample was assayed for human EGF and TGF-α concentrations by the standardized and validated ELISA Kit Quantikines (R&D Systems, Minneapolis, MN, USA) as per manufacturer’s instructions, whereas heparin binding-EGF (HB-EGF) and AR concentrations were measured with the standardized and validated DuoSet ELISA Development System (R&D Systems). This kit contains the basic components required for the development of sandwich ELISAs to measure human HB-EGF and AR, respectively. Briefly, the “capture antibody” for HB-EGF and AR was diluted at a working concentration of 2.0 and 0.4 μg/ml, respectively; 100 μl of the solution was placed in each well of a 96-well plate; the plate was sealed and incubated overnight at room temperature. The “detecting antibody” for HB-EGF and AR was diluted to a working concentration of 100 ng/ml in the Reagent Diluent (cat. #DY995, R&D Systems). Standards of recombinant human HB-EGF and recombinant human AR were reconstituted with the Reagent Diluent in order to obtain a seven-point standard curve as per manufacturer’s instructions. Measurements were performed by the microplate reader Multiskan Spectrum (Thermo Labsystems, Milan, Italy) set to 450 nm, with a wavelength correction set to 540 nm.
KRAS and BRAF mutational analysis
Sections of 10 μm thickness were obtained from formalin-fixed paraffin-embedded tissue samples. Tumor-rich areas were manually microdissected. Genomic DNA was extracted using the NucleoSpin Tissue kit (Macherey-Nagel GmbH, Düren, Germany) according to the manufacturer’s protocol and was kept at 4 °C before use.
KRAS codons 12, 13, and 61 and BRAF codon 600 mutations were investigated. High-resolution melting (HRM) analysis was performed as follows: PCR and HRM were consecutively performed on Rotor-Gene 6000™ (Corbett Research Ltd., Cambridgeshire, UK) in the presence of the fluorescent DNA-intercalating dye EvaGreen™ (Biotium Inc., Hayward, CA, USA). All reactions were performed in duplicate. Primers for mutational analysis were selected using the Primer3 software and were as follows: KRAS, 5′-TCATTATTTTTATTATAAGGCCTGCTG-3′ (forward), 5′-AGAATGGTCCTGCACCAGTAA-3′ (reverse); BRAF, 5′-TCCTTTACTTACTACACCTCAGAT-3′ (forward), 5′-AGTGGAAAAATAGCCTCAAT-3′ (reverse).
To confirm HRM analysis results, sequencing analysis was also performed on all samples. After HRM analysis, samples were purified with a QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA, USA) and submitted to cycle sequencing with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems Inc., Carlsbad, CA, USA) according to the manufacturer’s protocol and the same primers used in HRM analysis, but with 0.8 μmol/l in a final volume of 20 μl. The sequencing products were ethanol-precipitated before running on a 3130 Genetic Analyzer (Applied Biosystems Inc.). The sequencing data were visualized using Sequencing Analysis (Applied Biosystems Inc.) and were independently evaluated by two investigators. Each mutation was confirmed by sequencing a second independent PCR reaction.
EGFR and AR immunohistochemistry
EGFR IHC had been routinely performed with the EGFR pharmDx kit (Dako, Carpinteria, CA, USA), according to the manufacturer’s protocol. Specimens were considered EGFR positive when at least 1 % of malignant cells showed membranous staining. According to cetuximab label at the time of the study, only patients with EGFR-positive tumors were included. AR IHC was performed by the streptavidin–biotin method on 3 μm sections of primary tumors. A mouse monoclonal antibody against AR protein (Clone: 31221, R&D Systems, Inc.) was used at a 15-μg/ml dilution. 3-30-Diaminobenzidine and hematoxylin were used for color development and counterstaining, respectively. Immunostaining was considered positive if appropriate brown staining was detected in the cytoplasm of tumor cells. In the absence of a validated scoring system, we considered both the intensity of immunoreactivity and the percentage of stained cells, as previously described [13]. The percentage value of AR-positive cells for each tumor was evaluated calculating the percentage of immunoreactive cells in a total of 1,000 neoplastic cells. The intensity of the staining was classified according to a four-tier system: 0, no staining; 1, weak; 2, moderate; 3, strong. Scoring index was calculated by multiplying the intensity score by the percentage of stained cells, thus obtaining a final score ranging from 0 to 300.
Statistical analysis
Comparisons between concentrations at different time points were assessed by using the two-sided nonparametric Wilcoxon test. In the population of KRAS wild-type patients, the relation with response rate (RR), PFS, and overall survival (OS) was evaluated for all analyzed ligands. PFS was defined as the time from first treatment administration until disease progression or death. Patients who underwent secondary resection were censored at the time of surgery. OS was defined as the time from first treatment administration until death.
The receiver operating characteristics (ROC) analysis was performed for sensitivity and specificity of “1 h AR” cut-off value in predicting the RR. Survival curves were estimated using the Kaplan–Meier method, and the Cox proportional hazard model was adopted to estimate and test biological parameters for their association with survival. Results were expressed as hazard ratios (HRs) and relative 95 % confidence interval (95 % CI). For this purpose, variations of ligands’ concentrations were analyzed as continuous variables, in which the HR was referred to each increase of one percentage point. All statistical calculations were performed using the GraphPad Prism software package, version 5.0 (GraphPad Software Inc., San Diego, CA, USA) and R software, version 2.10.0 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Patients’ characteristics and clinical outcome
Forty-five patients (28:17, M/F), treated with cetuximab and irinotecan from January 2006 to July 2009, were included. From January 2009, only patients with KRAS wild-type disease have been treated. The main characteristics of the study population are summarized in Table 1. KRAS and BRAF were found mutated in 11 (24 %) and 5 (11 %) out of 45 cases. KRAS codon 12 and 13 mutations were reported in nine (81 %) and two (19 %) samples, respectively. Seven out of 34 patients with KRAS wild-type tumors achieved a partial response (RR 21 %). At a median follow-up of 22.1 months, in KRAS wild-type population, median PFS and median OS were 3.3 and 6.9 months, respectively. Paired plasma samples were available for 44 patients at d1, for 41 patients at d1-1hr and d15, for 38 patients at d15-1hr and d29, and for 23 patients at d57.
Modulation of EGFR endogenous ligands during treatment
EGFR ligands’ levels were differently modulated by the treatment with cetuximab and irinotecan.
AR basal levels and their variations significantly differ according to KRAS mutational status
In the overall population (Fig. 1a), AR levels rapidly increased (d1-1hr vs. d1, p = 0.013) and remained significantly higher than at baseline at subsequent time points (d15 vs. d1, p = 0.033; d29 vs. d1, p < 0.0001; d57 vs. d1, p = 0.0001).

Variations of amphiregulin (AR) plasma concentrations during the treatment with cetuximab and irinotecan, in the overall population (n = 45) (a) and in KRAS wild-type (n = 34) (b) and mutated (n = 11) (c) populations. Columns and bars, mean ± SD
AR basal levels were significantly higher among KRAS mutated patients, compared to KRAS wild-type ones (mean levels, 105.5 ± 39.7 vs. 41.0 ± 2.8 pg/ml; t test, p = 0.0073). A rapid increase in AR levels, reported in the overall population, was observed also in KRAS wild-type (d1-1hr vs. d1, p = 0.024; d15 vs. d1, p = 0.006; Fig. 1b), but not in the KRAS mutated subgroup (d1-1hr vs. d1, p = 0.250; d15 vs. d1, p = 0.695; Fig. 1c).
AR early increase at d1-1hr was significantly higher among KRAS wild-type patients, compared to KRAS mutated ones (mean variation, +43.5 vs. +6.0 %; t test, p = 0.023). AR tissue expression was measured by means of IHC in 22 primary tumors. No correlation of tissue and basal circulating levels of AR was reported (r = −0.04; p = 0.855).
EGF variations significantly differ according to KRAS mutational status
In the overall population (Fig. 2a), EGF levels significantly decreased straight after the administration of cetuximab both at d1 (d1-1hr vs. d1, p = 0.001) and at d15 (d15-1hr vs. d15, p = 0.0001). On the other hand, EGF significantly increased in the breaks between subsequent administrations (d15 vs. d1-1hr, p = 0.0001; d29 vs. d15-1hr, p = 0.040). EGF basal levels did not significantly vary according to KRAS mutational status (mean level, 53.8 ± 7.6 vs. 56.2 ± 8.2 pg/ml; t test, p = 0.868).

Variations of epidermal growth factor (EGF) plasma concentrations during the treatment with cetuximab and irinotecan, in the overall population (n = 45) (a) and in KRAS wild-type (n = 34) (b) and mutated (n = 11) (c) populations. Columns and bars, mean ± SD
The increase in EGF levels in the breaks between cycles, reported in the overall population, was observed also in the KRAS wild-type (d15 vs. d1-1hr, p < 0.0001; d29 vs. d15-1hr, p = 0.081; Fig. 2b), but not in the KRAS mutated subgroup (d15 vs. d1-1hr, p = 0.820; d29 vs. d15-1hr, p = 0.250; Fig. 2c). EGF increase at d15 was significantly higher among KRAS wild-type patients, compared to KRAS mutated ones (mean variation, +141.9 vs. +24.0 %; t test, p = 0.044).
TGF-α basal levels and their variations significantly differ according to BRAF mutational status
In the overall population (Fig. 3a), TGF-α levels increased, but not significantly, straight after the administration of cetuximab, both at d1 (d1-1hr vs. d1, p = 0.071) and at d15 (d15-1hr vs. d15, p = 0.055). TGF-α basal levels did not significantly vary according to KRAS mutational status (mean level, 32.0 ± 3.3 vs. 34.6 ± 5.9 pg/ml; t test, p = 0.707), but they were significantly higher among BRAF mutated patients, compared to BRAF wild-type ones (mean level, 54.3 ± 5.3 vs. 30.5 ± 3.0 pg/ml; t test, p = 0.008). Also in the KRAS wild-type subgroup, TGF-α levels were significantly higher among BRAF mutated patients compared to BRAF wild-type (mean level, 54.3 ± 5.3 vs. 28.8 ± 3.5 pg/ml; t test, p = 0.005). The increase in TGF-α levels, reported in the overall population, became significant in the BRAF wild-type subgroup (d1-1hr vs. d1, p = 0.046; d15 vs. d1, p = 0.018; d15-1hr vs. d1, p = 0.001; d29 vs. d1, p = 0.028; Fig. 3b).

Variations of transforming growth factor-α (TGF-α) plasma concentrations during the treatment with cetuximab and irinotecan, in the overall population (n = 45) (a) and in KRAS wild-type (n = 34) (b) and mutated (n = 11) (c) populations.Columns and bars, mean ± SD
Interestingly, TGF-α early increase at d1-1hr was significantly higher among BRAF wild-type patients, compared to BRAF mutated ones, both in the overall population (mean variation, +43.4 vs. −10.2 %; t test, p = 0.002) and in the KRAS wild-type subgroup (mean variation, +57.2 vs. −10.2 %; t test, p = 0.004).
HB-EGF levels were not significantly modulated during treatment
HB-EGF levels did not relevantly vary during the treatment, either in the overall population or according to KRAS or BRAF mutational status. Also HB-EGF basal levels were not significantly different according to KRAS or BRAF status.
EGFR ligands variations as predictors of outcome
In the KRAS wild-type population, AR and EGF early increases were associated with worse clinical outcome
As reported in Table 2, among KRAS wild-type patients, AR increase at d1-1hr vs. d1 was significantly higher among nonresponders, compared to responders (mean variation, +53.4 vs. +2.3 %; t test, p = 0.025). Also the EGF increase at d15 vs. d1 was significantly higher among nonresponders (mean variation, +80.0 vs. +9.5 %; t test, p = 0.017).
Similarly, as reported in Table 3, higher AR increases were significantly related to shorter OS (HR 1.853 [95 % CI 1.101–3.121], p = 0.020) and higher EGF increases at d15 were related, although not significantly, to shorter PFS (HR 1.286 [95 % CI 0.906–1.824], p = 0.159) and OS (HR 1.341 [95 % CI 0.931–1.932], p = 0.115).
Exploratory analysis of AR level at d1-1hr as potential predictor of clinical outcome by EGFR inhibition
Higher AR concentrations at d1-1hr were associated with worse survival both in KRAS wild-type (HR for PFS—1.006 [95 % CI 0.999–1.012], p = 0.058; HR for OS—1.010 [95 % CI 1.003–1.018], p = 0.0063) and in KRAS and BRAF wild-type populations (HR for PFS—1.025 [95 % CI 1.002–1.048], p = 0.035; HR for OS—1.029 [95 % CI 1.004–1.053], p = 0.020).
In an attempt to interpret “d1-1hr AR” as a dichotomized variable, an ROC analysis was performed, with the aim to determine the cut-off value, with “optimal” sensitivity and specificity, in predicting RR. The chosen cut-off value (d1-1hr AR plasma concentration of 42 pg/ml) had 83 % (95 % CI 36–100 %) sensitivity and 64 % (95 % CI 43–82 %) specificity, in predicting RR.
According to such a dichotomization, in the KRAS wild-type population, only 1 (6 %) out of 17 “d1-1hr AR high” patients (i.e., those patients with d1-1hr AR levels ≥ 42 pg/ml) achieved response, compared to 5 (36 %) out of 14 “d1-1hr AR low” patients (i.e., those patients with d1-1hr AR levels < 42 pg/ml; chi-square test, p = 0.067). Moreover, median PFS was 2.5 months in the “d1-1hr AR high” group vs. 4.4 months in the “d1-1hr AR low” group (HR 2.25 [95 % CI 1.04–4.87], p = 0.040; Fig. 4a) and median OS was 4.5 months in the “d1-1hr AR high” group vs. 8.4 months in the “d1-1hr AR low” group (HR 2.28 [95 % CI 1.02–4.83], p = 0.045; Fig. 4b).

Progression free survival (PFS) and overall survival (OS) according to d1-1hr amphiregulin (AR) concentration (cut-off 42 pg/ml) in KRAS wild-type (a and b, respectively) and in KRAS/BRAF wild-type populations (c and d, respectively). “d1-1hr AR high,” AR plasma levels ≥ 42 pg/ml; “d1-1hr AR low,” AR plasma levels < 42 pg/ml
Among the KRAS and BRAF wild-type populations, none out of 13 “d1-1hr AR high” patients achieved response, compared to 5 (42 %) out of 12 “d1-1hr AR low” patients (chi-square test, p = 0.015). Median PFS was 2.5 months in the “d1-1hr AR high” group vs. 5.3 months in the “d1-1hr AR low” group (HR 2.45 [95 % CI 1.26–8.02], p = 0.014; Fig. 4c), and median OS was 4.5 months in the “d1-1hr AR high” group vs. 9.9 months in the “d1-1hr AR low” group (HR 2.52 [95 % CI 1.24–8.37], p = 0.017; Fig. 4d).
In the KRAS wild-type population, AR, EGF, and HB-EGF levels at d57 are associated with response
In KRAS wild-type patients, EGF and HB-EGF levels at d57, when the radiographic assessment of response was performed, were significantly higher in nonresponders compared to responders. Also AR levels at d57 were higher, although not significantly, in nonresponders.
Discussion
In the last years, notable steps forward in the molecular characterization of mCRC have been taken. A growing amount of evidences underline that specific gene expression patterns are associated to well-known genetic features, such as KRAS and BRAF mutational status [14]. At the same time, the availability of anti-EGFR monoclonal antibodies for KRAS wild-type metastatic patients leads to the focus on the EGFR axis with the aim to disclose the mechanisms of intrinsic and acquired resistance to this class of drugs.
With specific regard to EGFR endogenous ligands, lower levels of AR and epiregulin mRNA were observed in KRAS mutated tumors, compared to KRAS wild-type ones, leading to hypothesize that in KRAS mutated tumors, EGFR ligands might contribute to the activation of the EGFR pathway to a lesser extent than in KRAS wild-type ones [9]. In our previous experience, we consistently observed that also BRAF mutated tumors express significantly lower levels of AR as detected by means of immunohistochemistry, compared to BRAF wild-type ones [15]. The same correlation of high tissue levels of AR and ER mRNA with KRAS and BRAF wild-type status was recently confirmed by the retrospective analysis of samples from the phase III COIN trial [16].
The potential role of tissue EGFR ligands in predicting the efficacy of anti-EGFR monoclonal antibodies was firstly suggested by the retrospective series by Khambata-Ford et al., evidencing an association of higher AR and ER mRNA levels with better clinical outcome [7]. More recently, similar findings were confirmed both by the post hoc analysis of the AIO CRC-0104 trial of CAPIRI plus cetuximab vs. CAPOX plus cetuximab [17] and by another retrospective series of patients receiving cetuximab in different lines of treatment [18]. The retrospective analysis of the phase III COIN trial revealed that higher levels of AR and ER are associated with better prognosis regardless of the administration of the anti-EGFR. Even more relevantly, high ER expression levels showed a predictive effect for the addition of cetuximab to first-line chemotherapy in KRAS wild-type patients.
In the present study, we planned to prospectively collect plasma samples from a clinically homogenous population of 5-fluorouracil-pretreated and irinotecan-refractory mCRC patients receiving salvage treatment with the association of cetuximab and irinotecan, as indicated by the first registrative trial of cetuximab by Cunningham and colleagues [11], in order to evaluate the modulation of plasma EGFR ligands according to KRAS and BRAF mutational status and to explore the potential predictive weight of these variations. The choice of the cetuximab plus irinotecan combination was preferred because the association of FOLFIRI and cetuximab is more commonly adopted as upfront treatment for mCRC patients, based on results of the phase III CRYSTAL trial [19].
We observed higher plasma levels of AR and TGF-α in patients with KRAS and BRAF mutated tumors, respectively. Apparently, these results are not consistent with previously reported findings about EGFR ligands’ tissue expression. A potential explanation may lie in the increased shedding activity of ADAM-17 metalloproteinase observed in KRAS mutated CRC cell lines that would be responsible for lower tissue and higher plasma levels of EGFR ligands [20].
Moreover, we observed that a rapid modulation of plasma EGFR ligands occurs following the exposure to cetuximab. Among patients with KRAS wild-type tumors, a smooth relationship was found between the early increase of AR levels and the HR for progression and death, thus suggesting that a more pronounced systemic reaction to the anti-EGFR is associated with a decreased probability of achieving benefit from the treatment. Another relevant finding of our study is the identification of a cut-off value of AR plasma levels (42 pg/ml) with reasonable sensitivity and specificity, allowing to interpret AR levels 1 h after the first administration of cetuximab as a dichotomic variable. Indeed, “d1-1hr AR high” patients showed significantly worst PFS and OS compared to “d1-1hr AR low” ones.
By a biologic perspective, our results are in line with preclinical findings by Mutsaers et al., showing a rapid elevation of human TGF-α in CRC cell lines’ conditioned medium following the exposure to cetuximab, and elevations in plasma TGF-α, AR, and epiregulin in mice treated with an anti-mouse EGFR monoclonal antibody. The same rapid increase was observed, after the administration of the anti-EGFR, in mice with or without CRC xenografts, thus supporting also a “host-mediated” contribution to this biologic response [21].
By a clinical perspective, our findings confirm previous results from the pharmacoproteomic analysis by Tabernero et al. [10], showing increased levels of AR, TGF-α, and EGF during treatment with the anti-EGFR in a subset of 45 patients enrolled in a phase I dose escalation trial of cetuximab monotherapy followed by FOLFIRI plus cetuximab. Nevertheless, no association with clinical outcome was previously reported.
Finally, interesting findings concern the modulation of circulating EGFR ligands occurring at the time of disease progression. In our experience, at the time of the radiologic assessment of response, higher levels of AR, HB-EGF, and EGF were found in nonresponding patients, suggesting a potential role of these ligands as a mechanism of acquired resistance to cetuximab and irinotecan. The opportunity to identify a “resistance-associated phenotype” could have major clinical implications. Circulating levels of EGFR ligands might contribute to the definition of this phenotype, since it seems that at the time of disease progression, KRAS wild-type patients present a plasma ligands’ profile that is superimposable to basal levels of KRAS mutated patients.
Although it is not currently known the mechanism by which the levels of physiological ligands of EGFR increase in plasma after administration of cetuximab, this phenomenon may be the possible basis of some mechanisms of resistance to this antibody. Panitumumab is a fully human immunoglobulin monoclonal antibody that binds the EGFR extracellular domain with high affinity and inhibits ligand-induced EGFR tyrosine phosphorylation, tumor cell activation, and tumor cell proliferation [22]. Moreover, recent data demonstrate that the antitumor activity of panitumumab correlates with its ability to penetrate into tumor tissue, saturate, occupy, and inhibit activation of EGFR [23]. Therefore, the use of drugs such as panitumumab, which exhibits a greater affinity and receptor occupancy, could be probably less affected by the increase of the EGFR physiological ligands and thus be less prone to the resistance phenomena that could potentially be associated with this increase. Further clinical studies in this direction will have to clarify this important issue.
The strongest point of the evaluation of EGFR ligands’ plasma levels lies in the opportunity to easily catch the dynamism of molecular changes occurring during the treatment, by a technically easy and minimally invasive assay. The understanding of molecular changes occurring early during the treatment, i.e., just 1 h after the infusion, may be useful not only to disclose the mechanism of action of anti-EGFR monoclonal antibodies, but also to optimize their use in the clinical practice. In our opinion, the present findings make 1hr AR levels a promising candidate as a potentially predictive pharmacodynamic marker to be investigated in validating studies. We are therefore conducting a confirmatory trial, based on a prospectively conceived formal hypothesis, in order to challenge this retrospective evidence.
References
Amado RG, Wolf M, Peeters M et al (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26:1626–1634
Bokemeyer C, Bondarenko I, Makhson A et al (2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 27:663–671
Karapetis CS, Khambata-Ford S, Jonker DJ et al (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359:1757–1765
Van Cutsem E, Kohne CH, Hitre E et al (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360:1408–1417
De Roock W, Claes B, Bernasconi D et al (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11:753–762
Loupakis F, Pollina L, Stasi I et al (2009) PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 27:2622–2629
Khambata-Ford S, Garrett CR, Meropol NJ et al (2007) Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 25:3230–3237
Harbison CT, Mauro DJ, Clark EA, Khambata-Ford S (2008) In reply. J Clin Oncol 26:2230–2231
Jacobs B, De Roock W, Piessevaux H et al (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27:5068–5074
Tabernero J, Cervantes A, Rivera F et al (2010) Pharmacogenomic and pharmacoproteomic studies of cetuximab in metastatic colorectal cancer: biomarker analysis of a phase I dose-escalation study. J Clin Oncol 28:1181–1189
Cunningham D, Humblet Y, Siena S et al (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351:337–45
Piantadosi S (2005) Translational clinical trials: an entropy-based approach to sample size. Clin Trials 2:182–192
Yonesaka K, Zejnullahu K, Lindeman N et al (2008) Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res 14(21):6963–6973
Popovici V, Budinska E, Tejpar S et al (2012) Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol 30:1288–1295
Cremolini C, Loupakis F, Ruzzo A et al (2010) Predictors of benefit in colorectal cancer treated with cetuximab: are we getting "Lost in TranslationAL"? J Clin Oncol 28:e173–e174, 4; author reply e175-176
Adam RA, Fisher D, Farragher S et al. (2012) Epiregulin (EREG) and amphiregulin (AREG) gene expression to predict response to cetuximab therapy in combination with oxaliplatin (Ox) and 5FU in first-line treatment of advanced colorectal cancer (aCRC). J Clin Oncol 30suppl: abstr 3516
Stintzing S, Jung A, Kapaun C et al. (2012) Ligand expression of the EGFR ligands amphiregulin, epiregulin, and amplification of the EGFR gene to predict for treatment efficacy in KRAS wild-type mCRC patients treated with cetuximab plus CAPIRI and CAPOX: analysis of the randomized AIO CRC-0104 trial. J Clin Oncol 30suppl: abstr 3519.
Pentheroudakis G, Kotoula V, De Roock W et al (2013) Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer 13:49
Van Cutsem E, Köhne CH, Hitre E et al (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360:1408–1417
Van Schaeybroeck S, Kyula JN, Fenton A et al (2011) Oncogenic Kras promotes chemotherapy-induced growth factor shedding via ADAM17. Cancer Res 71:1071–1080
Mutsaers AJ, Francia G, Man S et al (2009) Dose-dependent increases in circulating TGF-alpha and other EGFR ligands act as pharmacodynamic markers for optimal biological dosing of cetuximab and are tumor independent. Clin Cancer Res 15:2397–2405
Jakobovits A, Amado RG, Yang X et al (2007) From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol 25:1134–1143
Freeman DJ, McDorman K, Ogbagabriel S et al (2012) Tumor penetration and epidermal growth factor receptor saturation by panitumumab correlate with antitumor activity in a preclinical model of human cancer. Molecular Cancer 11:47
Acknowledgments
The authors thank Prof. Franco Bocci for his helpful suggestions. The present study has been partially supported by A.R.C.O. Foundation (Fondazione per la Ricerca e Cure in Oncologia) to AF and Associazione Italiana per la Ricerca sul Cancro (AIRC) to GB.
Conflict of interest
The authors have no conflict of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ethical statement
The authors state that the study has been approved by the local ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. In addition, informed consent has been obtained from all the participants involved.
Fotios Loupakis and Chiara Cremolini contributed equally to this study.
Rights and permissions
About this article
Cite this article
Loupakis, F., Cremolini, C., Fioravanti, A. et al. EGFR ligands as pharmacodynamic biomarkers in metastatic colorectal cancer patients treated with cetuximab and irinotecan. Targ Oncol 9, 205–214 (2014). https://doi.org/10.1007/s11523-013-0284-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-013-0284-7