
Abstract
Tumours with high 18F-FDG uptake values on static late PET images do not always exhibit high proliferation indices. These discrepancies might be related to high proportion of unmetabolised 18F-FDG components in the tissues. We propose a method that enables to calculate different 18F-FDG kinetic parameters based on a new mathematical approach that integrates a measurement error model. Six patients with diagnosed non-metastatic paragangliomas (PGLs) and six control patients with different types of lesions were investigated in this pilot study using 18F-FDG PET/CT. In all cases, a whole-body acquisition was followed by four static acquisitions centred over the target lesions, associated with venous blood samplings. We used an extension of the Hunter’s method to calculate the net influx rate constant (K H). The exact net influx rate constant and vascular volume fraction (K i and V, respectively) were subsequently obtained by the method of least squares. Next, we calculated the mean percentages of metabolised (PM) and unmetabolised (PUM) 18F-FDG components, and the times required to reach 80 % of the amount of metabolised 18F-FDG (T80%). A test–retest evaluation indicated that the repeatability of our approach was accurate; the coefficients of variation were below 2 % regardless of the kinetic parameters considered. We observed that the PGLs were characterised by high dispersions of the maximum standardised uptake value SUVmax (9.7 ± 11, coefficient of variation CV = 114 %), K i (0.0137 ± 0.0119, CV = 87 %), and V (0.292 ± 0.306, CV = 105 %) values. The PGLs were associated with higher PUM (p = 0.02) and T80% (p = 0.02) values and lower k 3 (p = 0.02) values compared to the malignant lesions despite the similar SUVmax values (p = 0.55). The estimations of these new kinetic parameters are more accurate than SUVmax or K i for in vivo metabolic assessment of PGLs at the molecular level.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
PET imaging using 18F-FDG is useful for grading tumours and assessing therapy or disease progression [8, 18]. The uptake of 18F-FDG is often characterised by calculating the standardised uptake value (SUV) from late static imaging (typically 60 ± 10 min after injection). However, the SUV is subjected to large variability, which compromises its use for inter- and intrapatient comparisons [9, 12]. Several normalisation schemes have been proposed to reduce its variability, but they do not account for the differences in 18F-FDG pharmacokinetics between individual patients. More importantly, these methods do not differentiate metabolised and unmetabolised 18F-FDG components within tumour regions [8]. Kinetic parameters can differentiate tumours with limited aggressiveness from benign lesions (e.g., low-grade liposarcomas vs lipomas) [6] or help to explain discordances such as high 18F-FDG uptake values in tumours with low proliferation indices.
The SUV assumes that the unmetabolised component of a radiopharmaceutical (e.g., in the blood within a tumour, in the intercellular spaces, and within the tumour cells themselves) is negligible; however, some authors have reported unmetabolised 18F-FDG components as high as 67 % [9]. The Patlak analysis enables the calculation of unmetabolised 18F-FDG, but this method has practical constraints (e.g., the acquisition of dynamic images and continuous arterial blood sampling) [16]. Simplified methods have been proposed as alternatives to the Patlak analysis to overcome the shortcomings of SUV (i.e., simplified kinetic analysis—SKA, simplified kinetic method—SKM), but these methods also suffer from their own limitations [11, 19]. SKA (i.e., Hunter’s method) neglects the unmetabolised fraction of 18F-FDG. SKM (i.e., Sundaram’s method) accounts for the unmetabolised 18F-FDG but is based on a rough estimation of the arterial input function. More recently, Hapdey et al. [10] extended the SKA method (ESKA) and significantly improved the accuracy and precision of K i estimates.
In the present study, we have calculated different 18F-FDG fractions and kinetic parameters based on a new mathematical approach that integrates a measurement error model. This approach was designed for routine use and is more elaborated than SKA but less time-consuming than the Patlak graphical approach. We focused the clinical evaluation of our approach on paragangliomas (PGLs) since these tumours often exhibit high 18F-FDG uptake values and low proliferation indices. Indeed, we hypothesised that these discrepancies are related to high proportions of unmetabolised 18F-FDG (e.g., unphosphorylated 18F-FDG) that are present in PGL tissue.
2 Materials and methods
2.1 Patients
Six patients with newly diagnosed PGLs and 6 control patients with benign or malignant lesions were included. The control group was composed of 3 benign (1 adrenal haematoma, 1 lung infection, and 1 schwannoma) and 3 malignant lesions (2 lung and 1 oesophageal carcinomas). In accordance with the local institutional guidelines, signed written informed consent was obtained from all patients prior to the participation.
3 18F-FDG PET/CT imaging
The patients fasted for a minimum of 6 h before 18F-FDG injection (4 MBq/kg), and scanning began approximately 60 min later (50–71 min). Blood glucose levels were within the normal range in all subjects at the time of the PET acquisitions. Three-dimensional images were acquired using a GE Discovery ST PET/CT hybrid scanner (General Electric Medical Systems). This scanner has an average axial 3D spatial resolution of 5.2 mm at 1 cm and 5.8 mm at 10 cm from the FOV centre and a maximum sensitivity of 9.3 cps/kBq. The axial and transverse FOV of this scanner are 15.7 and 70 cm, respectively.
The CTs were performed first and extended from the skull base to the upper thigh. The parameters for the CT were as follows: 140 kV, 64 mAs, DLP 388 mGy cm, and a 5-mm section thickness. The section thickness of CT scans matched the PET slice thickness. Immediately after the CT, a PET that covered the identical transverse field of view with an acquisition time of 3 min per table position (3D mode) was obtained.
Our first whole-body PET/CT was performed according to the current recommendations for cancer imaging [2] and helped us to precisely define the target hypermetabolic foci that were chosen for the following 4 additional list-mode acquisitions (3 min each every 5 min): t 1, t 2, t 3, and t 4.
The PET image datasets were corrected for random, scatter, and decay and iteratively reconstructed (OSEM algorithm) using the CT data for attenuation correction. Co-registered images were displayed on a workstation (Xeleris; GE Healthcare) with 3D representation and transaxial, coronal, and sagittal slices.
The 2D-ROIs were manually drawn on the 3 consecutives transaxial PET images surrounding the maximum intensity of the whole lesion. Each 2D-ROI covered at least two-thirds of the lesion surface. The same ROIs were used at each study time point (t 1, t 2, t 3, and t 4). For each ROI, the maximum activity concentration in (Bq/mL) and the maximum SUV (SUVmax) were measured.
Venous blood sampling was performed at 4 different time intervals: t 1, t 2, t 3, and t 4 which provided four measures of activity of 18F-FDG in the blood at time t j, noted, respectively, by C P,j for j = 1, 2, 3, and 4. The measurements of blood activity were taken using a Cobra Gamma Counter (Cobra II-Auto Gamma, Packard Instrument Co.). The 3-inch crystal configuration of this counter has a high sensitivity for detecting high-energy annihilation photons. Calibration was performed immediately before the sample measurements.
The counting error, which depends on the count rate, is approximately 1 % per 10,000 cps counted, the error on the volume measurement (<1 %) and the error on the counting efficiency, which should be estimated to be between 1 and 2 %, should be added to this the counting error.
4 Methods
To determine the unmetabolised fraction of 18F-FDG within the lesion, we considered the standard 3-compartment kinetic model [18]. The k 1, k 2, and k 3 transfer rate parameters characterise the transport between 2 extravascular compartments; k 1 measures the facilitated 18F-FDG transport from the blood into the tissue (a precursor compartment) per unit of tissue volume, k 2 measures the tracer transport from the precursor compartment back into the blood, and k 3 characterises the phosphorylation of 18F-FDG to 18F-FDG-6P (a metabolic compartment), which is assumed to be proportional to hexokinase activity. Our model assumes that, after phosphorylation, the radiotracer is irreversibly trapped in the tissue (k 4 = 0), which seems to be an appropriate approximation for various cancer models excluding hepatocellular carcinomas [15].
The unmetabolised 18F-FDG (e.g., the unphosphorylated 18F-FDG) includes the 18F-FDG located in the extracellular and the intracellular spaces. If FDG(t) denotes the tissue concentration of 18F-FDG (Bq/mL) in a target tissue and C P(t) denotes the concentration of 18F-FDG in the plasma, once the steady state is achieved, we have the well-known balance equation:
\({K_i}\int_0^t {{C_P}(\tau ){\text{d}}\tau }\) and VC P(t) represent estimations of the metabolised and unmetabolised 18F-FDG components, respectively. The parameter K i (min−1) is the so-called “net influx rate constant”; it is a composite rate of metabolised 18F-FDG extracted from the plasma and V (w/o unit), which is the vascular volume fraction in the tissue. The parameters \({K_i}\) and V are expressed in the following way:
and
From Eqs. (2) and (3), it follows that:
The primary objective was to determine \({K_i}\) and \(V\) to obtain estimations of both the metabolised and unmetabolised 18F-FDG components. These parameters depend on \({k_1}\), \({k_2}\), and \({k_3}\;\), but we do not need to calculate \({k_1},{k_2},{k_3}\) to estimate \({K_i}\) and \(V\). To identify these parameters with a method that would be feasible in clinical practice, we used a method that is an intermediate between SKA and Patlak graphical analyses. This method is based on a mathematical approach, integrates a model of the measurement errors and considers the arterial input function model \({C_{\text{P}}}(t)\) as proposed by the SKA method of Hunter et al. [11].
In the SKA method, \({C_{\text{P}}}(t)\) is modelled using a tri-exponential function as follows:
where b 1 , b 2, and b 3 are assumed to be equivalent for all patients and are determined from a set of patients for whom repeated blood sampling has been performed. For each individual patient, A 1 and A 2 are computed from the patient’s lean body mass and injected activity. A 3 is obtained by fitting the C P(t) model to a late blood sample. Equation 5 is then used to compute the area under the FDG(t) curve up to time t:
The \({K_{\text{H}}}\) index estimates the K i index based on the SKA method and is obtained by dividing the tumour FDG uptake by the AUC under the assumption that the distribution volume of 18F-FDG (V in Eq. 1) can be neglected.
In our study, \({K_H}\) was calculated using the b 1, b 2, and b 3 parameters provided in the work published by Hunter et al. [11]. As in the papers of Hunter et al., \({A_1} = {A_2}\) is the ratio of the injected dose to the blood volume, which is approximately equal to 70 mL per kilogram of lean body mass [10]. The constant A 3 was computed for each patient by fitting the \({C_P}(t)\;\;\) model to a late t 1 blood sample.
Because K i and V are independent of time, PET/CT images and venous blood samples can be performed at four different time intervals during the kinetic process. Given that a plateau phase can be observed to have small differences in terms of maximum activity concentrations, our model also accounted for the variability’s of the measurements of FDGj and C P,j as follows:
where \({\text{FD}}{{\text{G}}_j}\) is the maximum concentration averaged over the 3 2D-ROIs that were previously defined within the lesion at \({t_j}\). The function \({C_P}(t)\;\;\) is given by Hunter’s model and assuming that the experimental model error is given by ɛ and for j = 1, 2, 3, 4 by ɛ j . The random variables \(\bar \varepsilon\) and \(\;{\varepsilon_j}\) were distributed normally with mean 0 and with respective variance σ 2 and \(\;\sigma_j^2\) of the measurements at time t j . The error on the counting efficiency being estimated between 1 and 2 % allows to estimate σ 2 and at each acquisition time, and 3 values of maximum activity concentration were obtained which enables an estimate of the variance \(\;\sigma_j^2\;\) of FDG(t j ).
For each measurement of \({\text{FD}}{{\text{G}}_j}\) and C P,j , we drew 10,000 random samples of the \(\bar \varepsilon\) and ɛ j from a normal distribution with parameters 0 and σ 2 and 0 and σ 2 j , respectively. The number of random samples (n = 10,000) was selected based on the well-known Berry–Esseen inequality that specifies the rate at which convergence occurs by bounding the maximal error between the normal distribution and the true distribution of the scaled sample mean. With a convergence rate of \({n^{ - 1/2}}\), for n = 10,000, the error is less than 10−2. Next, given that \({K_i} \leqslant {K_H}\;\) because the calculation of K H neglects the unmetabolised 18F-FDG, we obtained the values of K i and V by minimising the following functional:
for 0 ≤ x ≤ K H and 0 ≤ y. That is, we obtain K i and V as follows:
Then, for each value \({\text{FD}}{{\text{G}}_{\text{j}}}\;{\text{and}}\;{C_{{\text{P}},{\text{j}}}}\) obtained by Eqs. 8 and 9, and after using the method of least squares which consists in minimising the functional shown in Eq. 10 (minimisation was performed using the classic Quasi-Newton method, which implemented in the software MATLAB we obtained 10,000 values for \({K_i}\) and \(V\;\) and then deduced their mean values.
Using the mean values previously obtained for \({K_i}\;{\text{and}}\;V,\) the estimations of the percentages of both the metabolised and unmetabolised 18F-FDG components at t were simple and are denoted by \({\text{PM(}}t )\,{\text{and}}\,{\text{PUM(}}t )\) , respectively:
It follows that, for any time T, the mean values of PM and PUM between 0 and T, which we were called μPM(T) and μPUM(T) are given, respectively by:
In the following, μPUM(60) will be noted PUM; therefore, PUM is the mean percentage of unmetabolised 18F-FDG between 0 and T = 60 mm (Table 2).
We were also able to calculate the time required to reach 80 % of the amount of metabolised 18F-FDG (\(T80\%\)). Note that \(T80\%\) characterises the rate of metabolism and is obtained as the single solution of the following equation:
The above equation (Eq. 16) can be easily solved numerically and a \(T80\%\) value can be estimated for each patient.
4.1 PGL confirmation
We focused the clinical evaluation of our approach on paragangliomas, which exhibit discrepancies between their low proliferation indices and high SUVs that are potentially explained by the contribution of unmetabolised 18F-FDG to the SUV values.
Histopathological analyses of the PGLs were considered the gold standard for the final diagnoses of PHEO/PGL and was obtained in 5 cases (cases 7–11). In the latter case, the diagnosis of PGL was made by a second imaging procedure using 3,4-dihydroxy-6-[(18)F]-fluoro-l-phenylalanine (18F-DOPA) which is considered as a specific tracer for head and neck PGL (HNPGL). 18F-FDOPA, and 123I-MIBG imaging were performed in HNPGLs and PHEOs, respectively. For 18F-FDOPA, patients fasted for 3 h before 18F-FDOPA injection (IASOdopa®, 4 MBq/kg). 18F-FDOPA PET/CT was performed without carbidopa pre-treatment. The PET emission scan started approximately 60 min after 18F-FDOPA injection. Three-dimensional images were acquired using a GE Discovery ST PET/computed tomography (CT) hybrid scanner (General Electrics Medical System). For 123I-MIBG scan, patients received at least 200 MBq intravenously (mean 220 MBq) and were evaluated at 24 h post-injection by planar whole-body scan (8 cm/min) using a dual head camera (ECAM, Siemens).
4.2 Statistics
All statistical tests were two-sided, nonparametric and performed using SPS 17.0 (SPSS Inc., Chicago, IL, USA). p values less than 0.05 were considered statistically significant. The agreement between K i and K H was evaluated using the intraclass correlation coefficient. Mann–Whitney tests were used for pairwise comparisons of the continuous measures between groups. Spearman’s rank correlation coefficients were calculated to assess the associations of the measures and kinetic parameters with the unmetabolised and metabolised 18F-FDG compartment.
5 Results
Table 1 shows the results of the evaluation of the robustness of the methodology. In the first part of Table 1, we performed a test–retest evaluation that involved repeating the calculations for each lesion 5 times. As shown in table, the repeatability was very good with coefficients of variation below 2 % regardless of the kinetic parameters considered.
Next, we assessed the robustness of our method across the time points \({t_1},{t_2},{t_3},{t_4}.\;\;\) For each patient, we computed the same parameters while considering the six 2 time points:
and the four 3 time points:
Note that for each time points of the calculation for K H becomes:
where \({t_{first}} = {t_1},{t_2}\;{\text{or}}\;{t_3}\) is the first measurement time considered as the case.
Then, the function to minimise becomes
where the set J is for 2 or 3 time points, respectively:
As shown in the second part of Table 1, the parameters were close to those obtained with all 4 time points. The maximum error for \({k_3}\) with respect to 4 time points was less than, 16 and 7.5 %, for 2 and 3 time points, respectively.
The results of the examination of the clinical feasibility of our method are given in Table 2, which shows the kinetic parameter estimates for all patients. Example images are shown in Figs. 1 and 2. The agreement between \({K_i}\;{\text{and}}\;V\) was excellent [intraclass correlation coefficient = 0.996, 95 % confidence interval (0.982–0.999)].

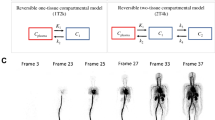
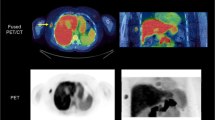
Cervical PGL. a 18F-FDOPA PET (maximal intensity projection (MIP)). b 18F-FDG PET (MIP). c 4 additional 3D acquisitions centred on the target lesion (18F-FDG PET MIP)

Lung adenocarcinoma. a 18F-FDG PET (MIP). b 4 additional 3D acquisitions centred on the target lesion (18F-FDG PET MIP)
This group of tumours was composed of 2 adrenal PGLs and 4 head and neck PGLs (HNPGLs). At the time of the study, all of the PGLs were considered as sporadic (based on the absence of a germline mutation in one of the susceptibility genes) and benign because malignancy is defined by the presence of metastatic lesions in which chromaffin cells are not typically present (i.e., lymph nodes, liver, lung, and bones). The Ki-67 proliferative indices were <1 % in all operated cases. The PGLs were characterised by high dispersions of the SUVmax (9.7 ± 11, coefficient of variation CV = 114 %), K i (0.0137 ± 0.0119, CV = 87 %), and V (0.292 ± 0.306, CV = 105 %) values and lower dispersions of the values of the new parameters \({k_3}\) (0.050 ± 0.007 min−1, CV = 14 %), \({\text{PUM}}\) (33.0 ± 3.9, CV = 12 %), and \(T80\%\) (38.34 ± 6.64, CV = 17 %).
The malignant lesions were characterised by higher values of SUVmax, \({k_3}\) , and metabolised 18F-FDG fraction compared to the control lesions (p < 0.05).
The SUVmax values were not significantly different between the PGLs and the malignant lesions (p = 0.44). The unmetabolised 18F-FDG fraction was found to be an important component of the 18F-FDG activities in the defined regions of all of the PGLs (median: 32.0 %) and was higher in these lesions than in the malignant lesions (median: 24.0 %). In the PGLs, high \({\text{PUM}}\) values were significantly associated with low k 3 (ρ = −0.99, p < 0.001) values. The PGLs were associated with higher \(T80\%\) (p = 0.02) values and lower k 3 (p = 0.02) values relative to the malignant lesions. Lastly, the PUM, k 3 and T80% values of the PGLs were similar to those of the benign lesions (p = 0.20).
6 Discussion
Standardised uptake value (SUV) is hampered by many simplifications and approximations, and the calculations of more reliable quantitative parameters would be of particular value to in vivo assessments of tumours at the molecular level [4, 5]. In the present study, we proposed a new methodology to calculate the metabolised and unmetabolised 18F-FDG fractions. We have evaluated this approach on PGLs because these lesions exhibit low proliferation indices and high uptake values, a finding that could be potentially attributable to the contribution of unmetabolised 18F-FDG to the SUV values.
In recent years, the use of PET/CT in PGL imaging has been increasing rapidly [20, 21]. These tumours, especially those associated with succinate dehydrogenase (SDH) mutations, are associated with high positivity on 18F-FDG PET [1, 20–23, 25]. These tumours often exhibit high SUVs despite their high degree of histological differentiation and low proliferation indices. Hypothetically, the activation of hypoxia signalling pathway has been invoked to explain the discordance between high 18F-FDG uptake and low proliferation (pseudo-hypoxia model) [26].
The present results suggest that PGLs are characterised by a relatively low 18F-FDG metabolic activity as expressed by the \({k_3}\), PUM, and \(T80\%\) values; contrasting with the high SUVmax values. Interestingly, PGLs with highly elevated SUVmax values were associated with higher \(T80\%\) values but relatively low \({k_3}\) values. From the pathophysiological standpoint, these findings might be related to the high uptake of 18F-FDG (via increased expression or activity of transporters) and a relatively low level of glycolytic activity. It is also notable that these new parameters exhibited lower CV values and should thus be more reliable than SUVmax and K i.
Despite the growing clinical relevance of 18F-FDG PET in oncology, little is known about the molecular determinants of tracer uptake in different types of tumours. Enhanced uptake and metabolism of glucose are frequently observed characteristics of most cancer cells and are associated with alterations to intrinsic energy metabolism that involve a shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis; this shift is referred to as the Warburg effect. The molecular mechanisms that underpin the metabolic reprogramming of cancer cells are complex and can involve adaptive responses to the tumour microenvironment such as hypoxia or mutations in enzymes or oncogenes that control cell metabolism [24].
PGLs associated with SDH or VHL genes mutations exhibit high positivity on 18F-FDG PET [1, 20–23, 25].
These results suggest that inactivation of the VHL and SDHx genes can upregulate specific HIF downstream targets (pseudohypoxia) and promote tumour growth, angiogenesis, and glycolysis. However, our results suggest that the high proportion of unmetabolised 18F-FDG fraction in PGLs might be related to lower rates of glycolysis than previously expected and to low proliferation rates. This supposition is consistent with the low \(T80\%\) (i.e., the rate of 18F-FDG phosphorylation) values observed in our PGLs, which are known to depend on hexokinase activity. \({K_{\text{i}}}\) might be elevated in some cases but is more likely to indicate 18F-FDG uptake via GLUT overexpression.
These results are consistent with experimental studies that have also failed to identify overexpression of HIF-1α and genes involved in glycolysis in most tumours [3, 7, 14, 17].
The high SUVs observed in PGLs are also currently not well explained or reflected by histopathological findings (differentiation, proliferation). These findings are also consistent with our preliminary results showing very low tumour 18F-FLT (fluoro-l-thymidine) uptake values despite very high 18F-FDG uptakes (manuscript submitted).
The unmetabolised 18F-FDG includes the 18F-FDG located in various compartments, including the extracellular spaces (in the blood and in the intercellular spaces) and in the cells (e.g., neuroendocrine cells and endothelial cells). In the present study, the PGLs were found to exhibit higher PUM and \(T80\%\) values than the malignant lesions. It is possible that \({\text{PUM}}\) might be influenced by genotype, but our cases had no mutations in one of the SDH genes.
We acknowledge several limitations to our study, including the small sample size, the absence of respiratory gating for three-dimensional PET of the thorax, and the lack of partial volume effect correction (lesion maximum diameter from 15 to 60 mm).
We assessed the robustness of this approach that was initially designed to consider 4 time points by using 3 or even 2 time points. Encouraging results were obtained (cf. Table 1). Another limitation of this study was the choice of 3 different 2D-ROIs surrounding the entire lesion to obtain the lesion maximum concentrations to derive the clinical variability of the FDG measurements, which yielded single \({K_i},V\;{\text{and}}\;{k_3}\) values for the entire lesion. This solution maximised the FDG measurement variability and thus might have increased the errors in the \({K_i}\;{\text{and}}\;V\) estimates. Further improvements are being developed to overcome these limitations. Our method was designed to be easily used in clinical routine (computation time below 1 min per lesion). Other potential improvements might include the obtention of the plasma time-activity curve from left-ventricle PET images [13] and a pixel-based generalisation of the methodology.
Another way of methodology improvement would be the use of regularised image reconstruction algorithm, recently implemented on the GE PETscans. Using this new algorithm, the image noise would be largely reduced and the \({K_i}\;{\text{and}}\;V\) estimation largely improved.
This study should be considered as a pilot case study and needs to be further evaluated in a larger study including more tumours with different genetic backgrounds.
7 Conclusion
In conclusion, the determination of the unmetabolised component can be of particular value to in vivo assessments of tumours at the molecular level. In this study, we proposed a new methodology for determining the metabolised and unmetabolised fractions of 18F-FDG. If our findings are prospectively confirmed in a larger patient population, they might provide a new approach for tumour characterisation by imaging and kinetic parameters should be evaluated as predictive biomarkers of malignancy.
References
Blanchet EM, Gabriel S, Martucci V, Fakhry N, Chen CC, Deveze A, Millo C, Barlier A, Pertuit M, Loundou A et al (2014) (18) F-FDG PET/CT as a predictor of hereditary head and neck paragangliomas. Eur J Clin Invest 44:325–332. doi:10.1111/eci.12239
Boellaard R, O’Doherty MJ, Weber WA, Mottaghy FM, Lonsdale MN, Stroobants SG, Oyen WJ, Kotzerke J, Hoekstra OS, Pruim J et al (2010) FDG PET and PET/CT: EANM procedure guidelines for tumour PET imaging: version 1.0. Eur J Nucl Med Mol Imaging 37:181–200. doi:10.1007/s00259-009-1297-4
Burnichon N, Vescovo L, Amar L, Libe R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J et al (2011) Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet 20:3974–3985. doi:10.1093/hmg/ddr324
Cheebsumon P, van Velden FH, Yaqub M, Hoekstra CJ, Velasquez LM, Hayes W, Hoekstra OS, Lammertsma AA, Boellaard R (2011) Measurement of metabolic tumor volume: static versus dynamic FDG scans. EJNMMI Res 1:35. doi:10.1186/2191-219X-1-35
Cheebsumon P, Velasquez LM, Hoekstra CJ, Hayes W, Kloet RW, Hoetjes NJ, Smit EF, Hoekstra OS, Lammertsma AA, Boellaard R (2011) Measuring response to therapy using FDG PET: semi-quantitative and full kinetic analysis. Eur J Nucl Med Mol Imaging 38:832–842. doi:10.1007/s00259-010-1705-9
Dimitrakopoulou-Strauss A, Strauss LG, Schwarzbach M, Burger C, Heichel T, Willeke F, Mechtersheimer G, Lehnert T (2001) Dynamic PET 18F-FDG studies in patients with primary and recurrent soft-tissue sarcomas: impact on diagnosis and correlation with grading. J Nucl Med 42:713–720
Favier J, Briere JJ, Burnichon N, Riviere J, Vescovo L, Benit P, Giscos-Douriez I, De Reynies A, Bertherat J, Badoual C et al (2009) The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 4:e7094
Freedman NM, Sundaram SK, Kurdziel K, Carrasquillo JA, Whatley M, Carson JM, Sellers D, Libutti SK, Yang JC, Bacharach SL (2003) Comparison of SUV and Patlak slope for monitoring of cancer therapy using serial PET scans. Eur J Nucl Med Mol Imaging 30:46–53. doi:10.1007/s00259-002-0981-4
Graham MM, Badawi RD, Wahl RL (2011) Variations in PET/CT methodology for oncologic imaging at US academic medical centers: an imaging response assessment team survey. J Nucl Med 52:311–317. doi:10.2967/jnumed.109.074104
Hapdey S, Buvat I, Carson JM, Carrasquillo JA, Whatley M, Bacharach SL (2011) Searching for alternatives to full kinetic analysis in 18F-FDG PET: an extension of the simplified kinetic analysis method. J Nucl Med 52:634–641. doi:10.2967/jnumed.110.079079
Hunter GJ, Hamberg LM, Alpert NM, Choi NC, Fischman AJ (1996) Simplified measurement of deoxyglucose utilization rate. J Nucl Med 37:950–955
Keyes JW Jr (1995) SUV: standard uptake or silly useless value? J Nucl Med 36:1836–1839
Li X, Feng D, Lin KP, Huang SC (1998) Estimation of myocardial glucose utilisation with PET using the left ventricular time–activity curve as a non-invasive input function. Med Biol Eng Comput 36:112–117
Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, Munoz I, Schiavi F, Montero-Conde C, de Cubas AA, Ramires R, Landa I, Leskela S et al (2010) Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol 24:2382–2391
Okazumi S, Isono K, Enomoto K, Kikuchi T, Ozaki M, Yamamoto H, Hayashi H, Asano T, Ryu M (1992) Evaluation of liver tumors using fluorine-18-fluorodeoxyglucose PET: characterization of tumor and assessment of effect of treatment. J Nucl Med 33:333–339
Patlak CS, Blasberg RG, Fenstermacher JD (1983) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 3:1–7. doi:10.1038/jcbfm.1983.1
Pollard PJ, El-Bahrawy M, Poulsom R, Elia G, Killick P, Kelly G, Hunt T, Jeffery R, Seedhar P, Barwell J et al (2006) Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab 91:4593–4598
Romer W, Hanauske AR, Ziegler S, Thodtmann R, Weber W, Fuchs C, Enne W, Herz M, Nerl C, Garbrecht M et al (1998) Positron emission tomography in non-Hodgkin’s lymphoma: assessment of chemotherapy with fluorodeoxyglucose. Blood 91:4464–4471
Sundaram SK, Freedman NM, Carrasquillo JA, Carson JM, Whatley M, Libutti SK, Sellers D, Bacharach SL (2004) Simplified kinetic analysis of tumor 18F-FDG uptake: a dynamic approach. J Nucl Med 45:1328–1333
Taieb D, Sebag F, Barlier A, Tessonnier L, Palazzo FF, Morange I, Niccoli-Sire P, Fakhry N, De Micco C, Cammilleri S et al (2009) 18F-FDG avidity of pheochromocytomas and paragangliomas: a new molecular imaging signature? J Nucl Med 50:711–717. doi:10.2967/jnumed.108.060731
Timmers HJ, Kozupa A, Chen CC, Carrasquillo JA, Ling A, Eisenhofer G, Adams KT, Solis D, Lenders JW, Pacak K (2007) Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol 25:2262–2269
Timmers HJ, Chen CC, Carrasquillo JA, Whatley M, Ling A, Havekes B, Eisenhofer G, Martiniova L, Adams KT, Pacak K (2009) Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 94:4757–4767
Timmers HJ, Chen CC, Carrasquillo JA, Whatley M, Ling A, Eisenhofer G, King KS, Rao JU, Wesley RA, Adams KT et al (2012) Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst 104:700–708. doi:10.1093/jnci/djs188
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Venkatesan AM, Trivedi H, Adams KT, Kebebew E, Pacak K, Hughes MS (2011) Comparison of clinical and imaging features in succinate dehydrogenase-positive versus sporadic paragangliomas. Surgery 150:1186–1193. doi:10.1016/j.surg.2011.09.026
Vicha A, Taieb D, Pacak K (2014) Current views on cell metabolism in SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 21:R261–R277. doi:10.1530/ERC-13-0398
Acknowledgments
This study was partly supported by a grant from Rotary Club of the Tholonet, in Aix-En-Provence. We wish to thank the patients who agreed to participate in the present study. The authors also wish to thank the technologists in the Department of Nuclear Medicine for their help in the management of the patients for this study. We lastly gratefully acknowledge Dr. I. Buvat for their help in improving this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Barbolosi, D., Hapdey, S., Battini, S. et al. Determination of the unmetabolised 18F-FDG fraction by using an extension of simplified kinetic analysis method: clinical evaluation in paragangliomas. Med Biol Eng Comput 54, 103–111 (2016). https://doi.org/10.1007/s11517-015-1318-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11517-015-1318-3