
Abstract
Methamphetamine (Meth) is an addictive psychostimulant abused worldwide. Ample evidence indicate that chronic abuse of Meth induces neurotoxicity via microglia-associated neuroinflammation and the activated microglia present in both Meth-administered animals and human abusers. The development of anti-neuroinflammation as a therapeutic strategy against Meth dependence promotes research to identify inflammatory pathways that are specifically tied to Meth-induced neurotoxicity. Currently, the exact mechanisms for Meth-induced microglia activation are largely unknown. NLRP3 is a well-studied cytosolic pattern recognition receptor (PRR), which promotes the assembly of the inflammasome in response to the danger-associated molecular patterns (DAMPs). It is our hypothesis that Meth activates NLRP3 inflammasome in microglia and promotes the processing and release of interleukin (IL)-1β, resulting in neurotoxic activity. To test this hypothesis, we studied the effects of Meth on IL-1β maturation and release from rat cortical microglial cultures. Incubation of microglia with physiologically relevant concentrations of Meth after lipopolysaccharide (LPS) priming produced an enhancement on IL-1β maturation and release. Meth treatment potentiated aggregation of inflammasome adaptor apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), induced activation of the IL-1β converting enzyme caspase-1 and produced lysosomal and mitochondrial impairment. Blockade of capase-1 activity, lysosomal cathepsin B activity or mitochondrial ROS production by their specific inhibitors reversed the effects of Meth, demonstrating an involvement of inflammasome in Meth-induced microglia activation. Taken together, our results suggest that Meth triggers microglial inflammasome activation in a manner dependent on both mitochondrial and lysosomal danger-signaling pathways.
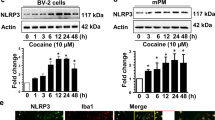
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methamphetamine (Meth) is a highly addictive psychostimulant and the second most widely used illicit drug worldwide, after cannabis (Degenhardt et al. 2010). Neurotoxic effects of chronic Meth abuse are detected by neuroimaging and psychological tests (Sekine et al. 2001; Chang et al. 2007). Chronic use of Meth leads to damage in dopaminergic and serotoninergic nerve terminals, which is associated with neuropsychiatric dysfunctions including deficits in episodic memory, decision making and mental illnesses like anxiety, depression, and psychosis (Scott et al. 2007; Glasner-Edwards et al. 2010). Ample evidence suggested that neuroinflammation played an important role in Meth-induced neurotoxicity (Loftis and Janowsky 2014). Studies have shown that Meth-mediated neurotoxicity is associated with upregulation of pro-inflammatory cytokines and chemokines (Krasnova and Cadet 2009; Loftis and Janowsky 2014). It has been shown that Meth treatment upregulates the expression of interleukin (IL)-1β, tumor necrosis factor α (TNF-α) and CCL2 in several brain regions including the striatum and frontal cortex (Flora et al. 2002; Nakajima et al. 2004; Kelly et al. 2012). As the resident immune cell in CNS system, microglia were found over-activated both in Meth-administrated animal models and patients (Thomas et al. 2004; Sekine et al. 2008). Because of the persistent neuroinflammation, the recovery often takes years (Qin et al. 2007; Gonzalez et al. 2014), Previous studies indicated that suppression of neuroinflammation significantly reduced the amount of self-administrated drug in mice and attenuated prime- and stress-induced relapse (Beardsley et al. 2010; Snider et al. 2013), suggesting that neuroinflammation might be a promising target for Meth abuse. However, the mechanisms by which Meth initiates the resident proinflammatory cascade are still largely unknown.
As microglia are key cellular mediators in neuroinflammatory processes, many early events of neurodegeneration are accompanied by microglial activation (Perry et al. 2010). Thus, the roles of microglial activation in Meth-induced neurotoxicity have been intensively studied (LaVoie et al. 2004; Thomas et al. 2004; Sekine et al. 2008; Friend and Keefe 2013). Mounting studies demonstrated that blocking microglial activation effectively reduced the Meth-associated neuronal damage and drug-seeking behavior (Thomas and Kuhn 2005a, b; Zhang et al. 2006; Hashimoto et al. 2007; Beardsley et al. 2010; Attarzadeh-Yazdi et al. 2014), supporting the central role of microglia in Meth-induced neuroinflammation. For this reason, the precise mechanisms underlying Meth activativation of microglia have been widely studied. Early characterization of Meth-induced microglial activation has demonstrated that many direct pharmacological effects associated with Meth, such as inhibition of the dopamine transporter (DAT), an increased release of dopamine (DA), and activation of D1 and/or D2 DA receptors, may not contribute to Meth-induced microglial activation (Thomas et al. 2004). On the other hand, the time-course, dose-response and pharmacological profiles of Meth-induced microglial activation suggest that microglia are not merely a secondary response to Meth-induced dopaminergic terminal damage (LaVoie et al. 2004; Thomas et al. 2004). Taken together, the cellular target that mediates the Meth-induced activation of microglia is still not identified.
It is well accepted that overactivated microglia produce and release proinflammatory cytokines, thus inducing collateral damage to neurons (Colton 2009). Thus, the most prominent feature of neurotoxic microglia is their secretion of proinflammatory cytokines like IL-1β. In our study, we primarily focused on identification of receptor that promotes production of the proinflammatory cytokine in response to Meth treatment. Previous study reported that Meth induces the release of damage-associated molecular patterns (DAMPs) such as high mobility group box-1 (HMGB1) that indirectly targets the microglia to upregulate IL-1β expression (Colton 2009). In a recent study focusing on inflammasome activation in Parkinson’s disease, the author discovered that after priming with LPS as first signal, the mitochondrial complex-1 inhibiting pesticide rotenone activated the inflammasome-induced IL-1β release and thus, promote the dopaminergic neurotoxicity (Sarkar et al. 2017). IL-1β is a well-recognized cytokine in the orchestration of CNS inflammation and is associated with elevated risk of drug dependence (Liu et al. 2009; Liu et al. 2011). On the one hand, alarming signals such as HMGB1 are suggested to play a priming role in neuronal stress-induced neuroinflammatory responses (Frank et al. 2015). On the other hand, chronic Meth abuser is more susceptible to various infections (Salamanca et al. 2014) and the potential disruptive effects of Meth on intestinal permeability are receiving more and more attention (Prakash et al. 2017). With the concurrent damage of BBB after Meth abuse, increased systematic level of endotoxins might get access to the brain (Northrop and Yamamoto 2015). Taken together, the sensitization of proinflammatory responses by these endo- or exogenous danger signals are primarily achieved by priming the NLRP3 inflammasome for subsequent immune challenges (Weber et al. 2015). The inflammasome is a multiprotein complex consisting of NOD-like receptors (NLRs), apoptosis-associated speck-like protein containing a CARD (ASC) and Caspase-1 (Walsh et al. 2014). Unlike other cytokines, IL-1β production is controlled by NF-κB and inflammasomes at the transcriptional and post-translational levels, respectively. The first signal upregulates the expression of pro-IL-1β, and the second signal leads to inflammasome activation, further processing and promoting the maturation of IL-1β. The activation of the inflammasome must be primed with toll-like receptor (TLR) signaling, which not only keeps NLRs from degradation but also increases the expression of pro-IL-1β. Caspase-1 is the converting enzyme that cleaves pro-IL-1β into the final active form (Walsh et al. 2014). Multiple endogenous danger signals, including lysosome rupture, ROS activation, and low intracellular K+, may be required for this process (Walsh et al. 2014).
In the present study, we investigated the primary effects of Meth on the production of proinflammatory cytokines on primary rat microglial cultures. Pathologically relevant concentrations of Meth fail to upregulate proinflammatory cytokines in mRNA levels but promote the maturation and release of IL-1β after LPS-induced pro-IL-1β increase. Thus, it is hypothesized that Meth potentiates IL-1β secretion and its activity by activating the inflammasome. Preventing this may preserve microglia and other nerve cells during early abstinence, which in turn should improve cognition and relieve withdrawal symptoms. The enhanced IL-1β maturation and secretion contributed to potentiated Meth-induced neurotoxicity, which suggests inflammasome as a potential target for future anti-neuroinflammation therapeutic drug development. A part of results was presented in an abstract form in the 21st Scientific Conference of the Society on NeuroImmune Pharmacology (SNIP) held in Miami, FL (April 22–25, 2015).
Materials and Methods
Chemicals
(+)-Methamphetamine (D-Meth), (−)-Methamphetamine (L-Meth) and LPS (from Escherichia coli 0111:B4) were purchased from Sigma-Aldrich (St. Louis, MO). Ac-YVAD-CMK, CA-074Me, and Mito-TEMPO were obtained from Enzo Life Sciences (Farmingdale, NY). All chemicals, unless otherwise specified, were from Sigma-Aldrich.
Animals
Pregnant female Sprague-Dawley (SD) rats used for experiments were purchased from Charles River Laboratories (Wilmington, MA). Animals were housed at constant temperature (22 °C) and relative humidity (50%) under a regular light-dark cycle (light on at 7:00 AM and off at 5:00 PM) with free access to food and water. All animal use procedures were strictly reviewed by the Institutional Animal Care and Use Committee (IACUC) of the University of Nebraska Medical Center (IACUC No. 13–069-10-EP).
Isolation and Culture of Microglial and Neuronal Cells
Microglia were prepared from the cerebral cortex of postnatal (0–1 day old) SD rats as described previously (Liu et al. 2012). Briefly, rat cortical tissues were dissected out in cold Hank’s Balanced Salt Solution (HBSS: Mediatech, Inc. Manassas, VA) and digested with 0.25% trypsin and 200 kunitz units/ml DNase (Sigma-Aldrich, St. Louis, MO) in 37 °C for 15 min (neuronal culture) or 30 min (microglial culture). The digested tissues were then suspended in cold HBSS and filtered through 100 μM and 40 μm pore cellular strainers (BD Bioscience, Durham, NC), respectively.
For the microglial culture, the isolated cells (25 × 106) were plated into T75 cm2 flasks in a high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 1× glutaMAX, 1% penicillin/streptomycin (Life Technologies, Grand Island, NY), and 300 ng/ml macrophage colony-stimulating factor (M-CSF) supplied by the Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center. After 10 days in culture, the flasks were gently shaked and detached cells were collected and seeded onto 35 mm2 (2.5 × 106 cells/dish) and 60 mm2 (7.5 × 106 cells /dish) culture dishes, and 12 well (1 × 106/well) or 96 well plates (0.4 × 106/well) based on the experimental requirements with M-CSF free DMEM. The suspensory glial cells were removed 1 h after seeding by changing the culture media. The resultant cultures were 98–100% microglia as determined by staining with anti-CD11b (Abcam, Cambridge, MA), a marker for microglia.
Primary cortical neurons were prepared from 19-day old embryonic rats. After physical dissociation, dissected cortical tissues were digested with 0.25% trypsin and DNase (200 Kunitz) in 37 °C for 15 min and then, filtered through 100 and 40 μM pore cellular strainers. Isolates were seeded onto poly-D-lysine-coated coverslips (0.2 × 106 cells/well in a 12-well plate) with plating medium (1% horse serum in neurobasal medium supplemented with 2% B27, 1% penicillin/streptomycin and 0.25% glutaMAX, all reagents from Life Technologies) for the first 6 h and exchange to culture medium for 10–14 days (plating medium without 1% horse serum and further supplemented with 20 μM 5-fluorouracil to reduce non-neuronal cell growth).
Quantitative PCR Analysis
The mRNA level of microglial pro-inflammatory cytokines were examined. Microglia was incubated with indicated concentrations of Meth for 12 h, with or without co-administration of LPS (100 ng/ml). Total RNA from microglia was extracted with TRIzol (Ambion, Carlsbad, CA). Two micrograms of RNA were reverse transcribed into complementary DNA (cDNA) with the SuperScript IV First-Strand Synthesis System (Invitrogen, Carlsbad, CA). cDNAs were amplified and quantified with the SYBR Green PCR Master Mix system (Applied Biosystems, Warrington, UK) and results normalized to GAPDH internal controls. PCR primers used were listed as follows: forward NLRP3 was TGG TCA AGG AGC ATC CAA GCA; reverse NLRP3 was AAG TGT TCA TCC TCA GGC TCA AA; forward IL-1β GAC CTG TTC TTT GAG GCT GAC A; reverse IL-1β AGT CAA GGG CTT GGA AGC AA; forward TNFα CGC CAC CGG CAA GGA; reverse TNFα GAC ATT CCG GGA TCC AGT GA.
Western Blotting
After priming with LPS for 6 h, the processing of pro-IL-1β and activation of capase-1 were analyzed after 12 h of Meth treatment. Microglia treated with a combination of Meth and LPS were lysed using RIPA buffer (Bio-Rad, Hercules, CA). Twenty microgram for total proteins was separated by 12% gel and transferred to nitrocellulose polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 3% BSA in tris-buffered saline (TBS) and incubated overnight at 4 °C with either mouse monoclonal antibody to IL-1β at a 1:500 dilution (R&D system, MN, USA), rabbit polyclonal antibody to caspase-1 at a 1:200 dilution (Santa Cruz, Dallas, TX) or anti-mouse β-actin monoclonal antibody (1:5000, Sigma-Aldrich). Washing buffer was TBS with 0.2% Tween (TBS-T). The secondary antibody was horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse antibody (1:10000, Jackson ImmunoResearch Laboratories, PA). Labeled proteins were shown by the Pierce enhanced chemiluminescence (ECL) system (Thermo Fisher Scientific, Waltham, MA).
Immunocytochemistry
Immunocytochemistry was conducted to quantify ASC aggregation as a readout of inflammasome activation. Microglia were seeded on coverslips in a 12-well plate at a density of 0.5 × 106 cells per well. After priming with LPS (100 ng/ml), antagonists were applied 1 h before Meth treatment. The lysosomal disruption was assessed by the immunocytochemistry staining of the lysosomal enzyme (Cathepsin B) after 24 h of Meth treatment. The cells were fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature. Then, the cells were blocked and permeabilized in PBS with 10% goat serum and 0.1% Triton X-100 for 30 min.
The primary antibodies used here including rabbit polyclonal antibody to ASC (Santa Cruz, CA, USA) and Cathepsin B (Sana Cruz, Dallas, TX) at 1:100 dilution. The microglia were identified by mouse monoclonal antibody CD11b in 1:500 dilution (Abcam, Cambridge, MA). The secondary antibodies used here were goat anti-rabbit Alexa 488 (1:1000) and Alexa 594 (1:1000) from ThermoFisher Scientific (Waltham, MA).
Fluorescent Dye Loading and Imaging
Fluorescent probes against ROS production, namely MitoSOX Red and H2DCFDA, were loaded onto the microglia that were treated with Meth for an additional 6 h after priming with LPS. MitoSOX Red and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (Life Technologies, Eugene, OR) were deployed to examine the intracellular ROS production. CM-H2DCFDA enters cells passively, and the acetate groups of the probe were cleaved by intracellular esterase leading to better cellular retention. After oxidation by reactive oxygen intermediates generated in response to Meth, the non-fluorescent H2DCFDA is converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF). The MitoSOX Red is a fluorogenic dye that is a highly selective indicator of ROS derived from mitochondrial in live cells. After treating with Meth for 6 h, the medium was removed and washed 3 times with pre-warmed PBS. The MitoSOX (2.5 μM) and CM-H2DCFDA (5 μM) working solutions were freshly made with pre-warmed DMEM medium and incubated on treated microglia at 37 °C for 30 min. The cells were then fixed with ice-cold 4% paraformaldehyde in PBS for 10 min and counterstained with DAPI. To quantify the results, the microglia were seeded onto a 96-well black plate in a density of 0.12 × 106/well and the treatment procedure mentioned above was then repeated. After loading with MitoSOX or CM-H2DCFDA, the intensities of specific fluorescent signals were evaluated by the microplate reader.
To assess the disruption of the lysosomal integrity of after 24 h of Meth administration, the specific molecular probe (LysoTracker) was applied onto microglia. The microglia seeded in a 12-well plate with coverslip were loaded with 50 nM LysoTracker Red DND-99 (Life Technology, Eugene, OR) in pre-warmed DMEM for 30 min. Cells were then washed 3 times and visualized by fluorescent microscopy.
TUNEL Staining
Neuronal apoptosis was assessed by In Situ Cell Death Detection Kit, Fluorescein (Roche Applied Science, Indianapolis, IN). Microglia was first treated with LPS or PBS for 6 h and then washed with pre-warmed DMEM (3 times, 10 min each). Different concentrations of Meth were added for additional 12 h and supernatants were collected for subsequent neuronal treatment. The media from treated microglia were diluted 5 times by basal neuronal medium and were transferred to the cultured neuron for 24 h. After fixation (4% paraformaldehyde) and permeabilizing (0.1% Triton X-100), neurons were then processed for TUNEL staining for 1 h at 37 °C and mounted using ProLong Gold antifade reagent with DAPI (Molecular Probes, Eugene, OR).
Cytokine Analysis by ELISA
In the experiments measuring secretion of IL-1β, microglia were primed with LPS and then stimulated with indicated concentration of Meth. After first 6 h priming with LPS (100 ng/ml), microglia were washed three times before further treatment of Meth. For detection of IL-1β release, the supernatant of Meth-treated microglia collected at 6 h, 12 h, and 24 h. The IL-1β in supernatants were detected using the IL-1β ELISA kit (R&D system, MN, USA). The experiments were performed following the manufacturer’s instructions. Briefly, the plates were coated with capture antibody overnight at room temperature and then, the filtered (0.2-μm strainer) BSA (0.1%) was used as the blocking reagent. The capture antibody-coated 96-well plate were incubated with collected supernatants for at least 2 h in room temperature. It followed by 2-h application of the detection antibody. Finally, the Streptavidin-HRP working solution was incubated for 20 min before substrate solution added to each well.
Measurement of Nitric Oxide (NO) Production
The concentration of nitrite was measured by the Griess Reagent System according to the manufacturer’s instruction (Promega, Madison, WI). After treatment of LPS and Meth, 50 μl aliquots of supernatant were collected from each group. Equal amounts of sulfanilamide solution and NED solution were added with collected supernatants for 10 min and 30 min separately. The absorbance of the final samples was measured on a plate reader with filters at 520 nm and 540 nm.
Caspase-1 Activity Measurement
Caspase-1 activity was measured by its colorimetric assay kit (RayBiotech, Norcross, GA). The labeled YVAD-p-nitroanilide (pNA) is the substrate of enzyme caspase-1, which allows this assay to detect the change of caspase-1 activity after treatment. The performance was followed by the manufacturer’s instructions. After cleavage from YVAD by the caspase-1 within cellular lysis of pretreated microglia, the light emission of chromophore pNA was quantified by a spectrophotometer at 405 nm.
Data Analyses
All data are expressed as mean ± S.D. unless otherwise indicated. Statistical analyses were performed by one-way ANOVA followed by post hoc analysis (indicated in each figure legend) in OriginPro 8.5 (OriginLab, Northampton, MA). A minimum p-value of 0.05 was chosen as the significance level for all tests. The densities of target western blot bands were quantified by AlphaView (ProteinSimple, San Jose, CA) and standardized by β-actin band density. In the section using a specific fluorescent probe against lysosomes (LysoTracker Red DND-99), 9 fields under the fluorescent microscope were taken, and all cellular fluorescent intensities were quantified by Image J (Schneider et al. 2012). The intensities of all individual cells in each field were averaged and transformed to fold changes against a control group. As for the activity of caspase-1 and NO production assay, the arbitrary absorbance of treated groups was also displayed as fold change against the control group. To quantify the neuronal apoptosis induced by Meth, 6 different fields under 40× objective were captured for each treatment group. The percentage of apoptotic neurons was analyzed by the number of TUNEL-positive cells normalized by the total DAPI signals. All experiments were performed in triplicate unless otherwise specified.
Results
Transcriptional Analysis of Proinflammatory Cytokines after Meth Treatment
To investigate if proinflammatory cytokines were upregulated in isolated microglia treated with Meth, we analyzed the production and secretion of IL-1β and TNFα by qPCR and ELISA. After Meth treatment at different concentrations, the expression levels of proinflammatory cytokines (IL-1β and TNFα) exhibited no significant change at transcriptional levels (Fig. 1a, d). In parallel, the secreted IL-1β in collected supernatants collected from Meth-treated microglia (both in dextrorotary and levorotatory form) was undetectable while it was upregulated in supernatants of the LPS-treated group (Table 1). To evaluate the effects of Meth on pre-existing inflammation, the transcriptional levels of proinflammatory cytokines in LPS- and Meth-treated microglia were analyzed. The results showed that Meth treatment did not further upregulate the production of IL-1β and TNFα after LPS stimulation (Fig. 1b, e). As our study primarily focuses on the inflammasome-induced IL-1β signaling pathway, we next examined IL-1β production at different time points. As shown in Fig. 1c, the effect of Meth potentiation on LPS-induced IL-1β production was marginal significant (p = 0.07) at 6 h, but not at 12 and 24 h. These results indicate that Meth, treated alone or combined with LPS stimulation, did not alter the production of proinflammatory cytokines.

Meth treatment did not upregulate production of proinflammatory cytokines. The transcriptional levels of IL-1β and TNFα were analyzed by qPCR after treatment of Meth on isolated microglia. Effects of different concentrations of Meth were measured with (b, e) or without (a, d) priming signal (100 ng/ml LPS). The potentiation effects of Meth (50 μM) on LPS (100 ng/ml) was also measured at different time points (c)
Meth Enhancement of Processing and Release of IL-1β
IL-1β processing and release are regulated by the inflammasome signaling. It is well-documented that full activation of the inflammasome requires the first priming signal to upregulate their major components and the primary substrate. As shown in Fig. 1a, Meth appears not function as the first signal for inflammasome activation. Thus, Meth may work as the second signal for inflammasome activation and stimulate the processing and release of IL-1β. Therefore, we examined the maturation of IL-1β indicated by the increase of its smaller-sized activation form and its subsequent release in Meth-treated microglia. After priming with LPS (classical first signal for inflammasome activation) for 6 h, the secretion of IL-1β was significantly potentiated along with increasing concentrations of Meth (Fig. 2a). To determine whether this potentiation is receptor-mediated, a levorotary form of Meth (L-Meth) was introduced into experiments as a comparison. The potentiation effects of the dextrorotary form of Meth (D-Meth) started at the concentration of 6 μM and peaked at 50 μM for a 2.6-fold increase after priming with 100 ng/ml LPS (Fig. 2a). Unlike D-Meth, the L-Meth is less potent, with significant increase of IL-1β release only detected in 50 and 150 μM of Meth administration and peaking at 50 μM for the 1.5-fold increase (Fig. 2b). However, both enantiomers showed a dose-dependent manner of potentiation on LPS-induced IL-1β release (Figs. 2a and b). To better characterize the potentiation effect of Meth on LPS-induced IL-1β release, the time course experiment of D-Meth was performed to determine the best time point for subsequent studies. As shown in Fig. 2c, the maximal effect (2.1-fold increase compared to the LPS group) of D-Meth was reached at 12 h. Thus, this time point was selected for the investigation on Meth-induced inflammasome activation. The following studies on IL-1β maturation also confirmed that Meth promotes the cleavage of pro-IL-1β into its smaller-sized activation form in a dose-dependent manner (Fig. 2d). While the production of pro-IL-1β was not significantly changed after 12 h treatment of Meth, the 17-kD form of IL-1β was significantly increased. Consistent with the ELISA results, Meth promoted the processing of IL-1β at 6 μM and reached the maximal effect at 50 μM (1.7-fold increase compared to control). These results demonstrated that the processing and release of IL-1β were promoted by Meth after priming with LPS, which indicates a possible role of inflammasome activation in Meth-induced potentiation.

Meth treatment potentiated LPS-induced IL-1β processing and release. Microglia were pretreated 6 h with LPS (100 ng/ml) and then incubated 12 h with different concentrations of D-Meth (a) or L-Meth (b). Alternatively, after 6 h pretreatment of LPS, cells were incubated with Meth (50 μM) for indicated times (c). The supernatants were collected, and concentrations of secreted IL-1β were measured by ELISA. The optimized time point (12 h) and concentration (50 μM) of Meth treatment were applied on cultured microglia. The production and processing of IL-1β were detected by western blot (d). After standardized with β-actin, the densitometry of bands was analyzed and quantified, as shown in bar graphs (d). * p < .05, ** p < .01, *** p < .001 vs LPS
Meth Activation of Inflammasome
The inflammasome, a molecular platform, is comprised of the sensing molecule NLRP3, the adaptor ASC protein, and the executive enzyme caspase-1. The primary direct evidence against inflammasome activation is oligomerization of the ASC protein and auto-cleavage activation of caspase-1. To investigate whether Meth promotion of IL-1β maturation by activation of the inflammasome, we examined the processing of pro-caspase-1 by western blot and the visualized oligomerization of ASC by immunofluorescent staining. Also, the caspase-1 inhibitor was introduced to prove that the Meth-induced IL-1β activation was specifically mediated by caspase-1 activation. The functional mature form of caspase-1 (10-kD band in Fig. 3a) was significantly upregulated with Meth treatment while the expression levels of pro-caspase-1 exhibited no change (Fig. 3a). The functional cleaved form of caspase-1 was significantly increased with 20 μM Meth treatment and reached 1.8-fold increase with 50 μM Meth. Likewise, the colorimetric assay showed a significant increase in caspase-1 activity in a similar pattern (Fig. 3c). As a pivotal component of the inflammasome, ASC proteins aggregate after NLRP3 activation to help assemble the inflammasome platform. In our experiments, the ASC protein formed aggregates that appear as “speck”-like spots near the nuclear periphery, indicating inflammasome formation after Meth treatment (marked by arrows in Fig. 3d). No ASC aggregation was detected in either the control or LPS groups and ASC accumulated only after treatment with Meth. The number of ASC aggregates also reached its highest level in the 50 μM Meth-treated group. Consistent with the results of IL-1β processing, the maturation of IL-1β and caspase-1 did not increase further at the highest concentration of Meth (150 μM). Thus, Meth (50 μM) was employed for the subsequent studies. To ensure that the Meth-induced IL-1β maturation is specifically mediated by inflammasome activation, the selective caspase-1 inhibitor Ac-YVAD-CMK at 10 μM (Walker et al. 1994; Garcia-Calvo et al. 1998) was applied 1 h before Meth treatment. As shown in Fig. 3b, the pretreatment of caspase-1 inhibitor significantly reduced the Meth-induced processing of IL-1β while the expression levels of pro-IL-1β unchanged. These results demonstrated that Meth-induced inflammasome activation by aggregation of the ASC protein and specific activation of caspase-1.

Meth treatment induced inflammasome activation in a dose-dependent manner. The cleavage and activation of caspase-1 were measured by western blot and quantified by colorimetric assay. The cultured microglia were primed with LPS 100 ng/ml for 6 h and treated with different concentrations of Meth for another 12 h. To evaluate the magnitude difference of the precursor and cleavage form of caspase-1, the densitometry of 45 kD pro-caspase-1 and 10 kD caspase-1 were quantified separately, which are shown in associated bar graphs (a). The enzyme activity of caspase-1 after incubation of Meth was measured by caspase-1 colorimetric assay (c). After 6 h priming of LPS, the caspase-1 inhibitor Ac-YVAD-cmk (10 μM) was applied 1 h before Meth treatment and the processing of IL-1β was measured after another 12 h of Meth (50 μM) treatment (b). As another indicator of inflammasome activation, the change of distribution of ASC protein was visualized by immunofluorescent microscope (d). The formation of ASC oligomers was detected in the cytoplasm and marked by red arrows (d). Blue: DAPI; Green: CD11b; Red: ASC. * p < .05, ** p < .01, *** p < .001 vs LPS. # p < .05, ### p < .001 vs Ctrl. $$ p < .01 vs LPS + Meth 50 μM. Scale bar, 100 μm
Identification of Danger Signaling-Associated Molecular Pathways Leading to Meth-Induced Inflammasome Activation
The NLRP3 inflammasome activation may occur through multiple cellular danger signaling pathways including the induction of ROS, lysosomal disruption and cathepsin B release, potassium efflux, and calcium mobilization. Accumulating evidence suggests that Meth application could activate the mitochondrial ROS production and disrupt the normal lysosomal function (Cubells et al. 1994; Wu et al. 2007; Tian et al. 2009; Potula et al. 2010; Shiba et al. 2011; Funakoshi-Hirose et al. 2013; Mashayekhi et al. 2014). To identify the inflammasome-related upstream molecular events induced by Meth, specific inhibitors against mitochondrial ROS and lysosomal pathways were tested to examine if Meth induces IL-1β release via specific signaling pathways. After pretreatment of glial cells with specific inhibitors, immunofluorescent staining of the ASC protein and measurement of secreted IL-1β were performed on Meth-treated microglia. As shown in Fig. 4a, the cathepsin B inhibitor CA-074Me at 10 μM (Hill et al. 1994) totally reversed the Meth-induced ASC protein oligomerization (Fig. 4a). However, aggregation of the ASC protein was only partially blocked with pretreatment of the mitochondrial ROS inhibitor Mito-TEMPO at 100 μM (Liang et al. 2009) (Fig. 4a). To ensure the specificity of these inhibitors, the dose-dependent response bar graphs on Meth-induced IL-1β release were generated (Fig. 4b). To test if Meth potentiaton of IL-1β release through upregulation of major components of the inflammasome, we examined the transcriptional levels of NLRP3 after application of LPS and Meth. LPS, as the classical first signal, successfully upregulated the expression of NLRP3, while Meth had no significant effects on alteration of the NLRP3 production (Fig. 4c).

Meth-induced inflammasome activation was inhibited by lysosomal and mitochondrial ROS-specific inhibitors. Punctate ASC immunoreactivity was detected in the cytoplasm of Meth (18 μM) treated microglia (a). Microglia pretreated with lysosomal cathepsin B inhibitor (CA-074Me 10 μM) and mitochondrial ROS inhibitor (Mito-TEMPO 100 μM) exhibited minimal ASC immunoreactivity (a). Blue: DAPI; Green: CD11b; Red: ASC. Isolated microglia were pretreated with different concentrations of inhibitors before LPS and Meth application; the secreted IL-1β were quantified by ELISA (b). The transcriptional level of NLRP3 was measured by qPCR on Meth-treated microglia with or without priming signal (c). *** p < .001 vs LPS; # p < .05, ## p < .01, ### p < .001 vs LPS + Meth 50 μM. Scale bar, 50 μm
Production of Mitochondrial ROS after Meth Treatment
Mitochondria are a major source of inflammasome-activating ROS (Zhou et al. 2011). To investigate if Meth-induced ROS formation primarily locates at the mitochondria, a molecular probe that distinguishes the source of ROS was employed. After loading with the specific molecular probe, the fluorescent microscope was used to take images, and the microplate reader was used to quantify the results. As a total endogenous ROS indicator, H2DCFDA fluorescent intensities increased after LPS stimulation. Meth application further activated the endogenous ROS system evidenced by elevated H2DCFDA fluorescence in a dose-dependent manner (Fig. 5a and c). Consistently, the mitochondria-specific probe Mito-SOX also indicated a further increase of mitochondrial ROS after Meth treatment (Fig. 5b and e). The accumulated red signals indicating mitochondrial ROS are primarily localized in the cytosol but not in the nucleus as marked by DAPI, suggesting that the probes target the mitochondria. The peak effect of Meth on total ROS formation reach a 1.7-fold increase compared with the LPS group. Matching this magnitude, the Meth-induced generation of mitochondrial ROS peaks at 1.6-fold compared to the LPS group. In addition to ROS, reactive nitrogen species (RNS) are also found mediating the Meth-induced neurotoxicity (Quinton and Yamamoto 2006). To test if the activation of RNS is also incorporated into the Meth-induced cellular stress system, the nitrites secreted in the supernatants of Meth-treated microglia were measured and quantified as shown in Fig. 5d. There is no significant change of the nitrite level after Meth treatment.

Meth treatment induce Mitochondrial ROS generation but not NO production. The total ROS and mitochondrial ROS are detected by specific fluorescent molecular probe. The change of the fluorescent intensity was visualized by microscope and quantified by the microplate reader. Isolated microglia were primed with LPS 6 h and then incubated with indicated concentrations of Meth. Fluorescent images of DCFDA green signals (a) and MitoSOX Red signals (b) were taken in microglia after 12 h of Meth treatment. The quantitative analyses of DCFDA and MitoSOX signals were exhibited in (c) and (e). NO productions in harvested supernatants were measured using Griess reagent system (d). * p < .05, ** p < .01, *** p < .001 vs Ctrl. # p < .05, ## p < .01, ### p < .001 vs LPS. Scale bar, 100 μm
Permeabilization of Lysosomal Membrane in Meth-Treated Microglia
Another inflammasome-related pathway identified is the Meth-induced cathepsin B release. To further investigate the mechanism of lysosome disruption and subsequent content release after Meth application, lysosomal membrane permeabilization (LMP) was assessed in this cellular model. The assessment of lysosomal integrity was achieved by measuring the retention rate of LysoTracker and detecting the intracellular distribution of lysosomal protease cathepsin B. LysoTracker-labeled lysosome in the control group revealed a cytoplasmic punctuate structure, but with increasing concentrations of Meth, fluorescence intensity and the number of punctuates apparently decreased (Fig. 6a). LPS administration significantly increased the signals of LysoTracker while additional treatment of Meth reduced the signals in a dose-dependent manner (Fig. 6a). Another sign of LMP is the translocation of soluble lysosomal protease from the lysosomal lumen to the cytosol. Immunofluorescence staining revealed a redistribution pattern of cathepsin B after Meth application, marked by the loss of punctuating shape and diffusion of stain throughout the cellular cytosol (Fig. 6b). However, as the cellular marker of the lysosome, the fluorescent intensities of lysosomal-associated membrane protein 1 (LAMP-1) did not change with Meth treatment (Fig. 6b).

Meth treatment disrupts the lysosomal integrity. Lysosomal integrity was measured by LysoTracker (Red). The fluorescent intensity was quantified and exhibited in (a). Colocalization of cathepsin B and lamp-1 was assessed by fluorescent microscopy (b). *** p < .001 vs Ctrl; ### p < .001 vs LPS. Panel A, scale bar, 100 μm. Panel B, scale bar, 50 μm
Enhancement of LPS-Induced Microglial Neurotoxic Activity by Meth Treatment
Previous results have demonstrated that Meth could potentiate LPS-induced IL-1β release by activation of the inflammasome. The microglia-neuron co-culture system was used to determine whether Meth application potentiated LPS-induced neurotoxicity. After Meth treatment on LPS-primed microglia, the collected supernatants were used in experiments for examining neurotoxicity. To detect neuronal apoptosis, TUNEL staining was performed 12 h after complete removal of neuronal culture medium and replaced with the microglia culture medium at a dilution ratio of 1:5. Because previous studies indicate that Meth alone could induce neuronal apoptosis, we included the group of Meth alone as a comparison (Jayanthi et al. 2004). The percentage of TUNEL-positive cells was significantly increased after Meth treatment (Fig. 7a). Although the administration of LPS (100 ng/ml) and Meth (50 μM to 150 μM) each alone also increased the percentage of the apoptotic neurons, application of LPS and Meth in combination further significantly potentiated the proapoptotic effects (Fig. 7a). There was 1.8-fold increase compared to LPS alone and 1.5-fold increase compared to Meth alone. Pre-treatments of cultured microglia with inhibitors against caspase-1 activity (YVAD), cathepsin B (CA-074Me), and mitochondrial ROS (Mito-TEMPO) each alone significantly reduced the percentage of apoptotic neurons in comparison to the LPS + Meth 50 μM group (Fig. 7b), once again supporting Meth activation of inflammasome even though the inhibitors did not totally reverse the Meth-associated apoptotic effects.

Meth treatment potentiated LPS induced neurotoxicity and blocked by inflammasome inhibitors. Neuronal apoptosis was analyzed by combined TUNEL/DAPI staining, and quantification of apoptotic neurons was performed by normalization of TUNEL-positive cells to a total number of DAPI-positive cells. The apoptosis induced by incubation with supernatants of cultured microglia treated with indicated concentrations of Meth. The increased apoptotic neurons were visualized by green TUNEL-positive signals that were notably observed in LPS + Meth treatment group (a). The percentage of apoptotic neurons were counted and statistically analyzed. The quantified results were exhibited in the bar graph of panel A. The blockage effect of inflammasome inhibitors on Meth-induced neuronal apoptosis were examined and quantified in panel B. After priming with LPS for first 6 h, the inhibitors were applied 1 h before additional Meth treatment. The collected supernatant was transferred to cultured neuron and TUNEL-staining were performed. * p < .05, ** p < .01, *** p < .001 vs Ctrl. # p < .05, ## p < .01, ### p < .001 vs LPS. && p < .01, &&& p < .001 vs LPS + Meth 50 μM. Scale bar, 100 μm
Discussion
Although neuroinflammatory responses marked by microgliosis and astrogliosis have been observed in Meth abusers (Sekine et al. 2008; Robson et al. 2014), the mechanisms by which Meth directly contributes to the initiation and progression of the microglia-mediated neuroinflammation remains unclear. As a strong addictive psychostimulant, Meth abuse induces not only a strong euphoric effect leading to addiction but also affects innate and adaptive immunity, thereby causing neurotoxic activity (Loftis and Janowsky 2014). In this study, we evaluated effects of Meth on proinflammatory cytokine production in microglial cells. The results showed that Meth activated endogenous danger signal pathways, promoting inflammasome-mediated IL-1β maturation and secretion. Meth caused microglial lysosome disruption and ptomoted mitochondrial ROS production, both of which activated the inflammasome-induced IL-1β maturation and secretion at post-translational level. Blockage of Meth-associated inflammasome upstream danger signals by specific inhibitors, CA-074Me or Mito-TEMPO, significantly inhibited inflammasome activation and IL-1β secretion. These results suggest that Meth potentiates the microglia-induced neurotoxicity by increasing the production and release of inflammasome-mediated IL-1β.
To reflect the conditions in human subjects, the concentrations of Meth used in this study were based on the biodistribution (BD) profiles from previous studies (Melega et al. 2007). The Meth levels in the circulating blood of abusers were 2.0 μM on an average, with the highest of 11.1 μM (Melega et al. 2007). The Meth levels in the brain of abusers can be further estimated according to the tissue-to-serum ratio of Meth in animals, with an average of 9.7 for brain distribution (Riviere et al. 2000). According to this distribution ratio, the dose range of Meth was 6–150 μM. Thus, the Meth concentrations used in this study were within the aforementioned range.
Proinflammatory cytokines are important mediators of microglia-induced neurotoxicity in many neurodegenerative disorders (Lull and Block 2010). To examine Meth effects on the transcription of proinflammatory cytokines, the mRNA levels of IL-1β and TNFα were detected after Meth administration at different concentrations and time courses. LPS was used as a positive control to rule out the possibility of microglial anergy. When tested alone, Meth had no significant effects on the alteration of mRNA levels of IL-1β and TNFα in cultured rat microglia. These results were, on the one hand, consistent with a previous study that failed to detect any Meth-induced direct proinflammatory responses in the cultured microglia. However, it was, on the other hand, inconsistent with the in vivo results that up-regulated proinflammatory signals were observed after Meth administration (Frank et al. 2016). The possible explanations for the discrepancy are that Meth promotes the alarming signals released from neurons, resulting in upregulation of proinflammatory signaling in microglia. The increased infection susceptibility and the altered BBB structure after Meth treatment further complicated the situation. Combined with our results, Meth administration may potentiate the existing inflammation by the unknown mechanism.
For this reason, we performed experiments to investigate whether Meth treatment could enhance the pre-existing inflammatory stimulation in cultured microglia. Our results showed that Meth treatment enhanced the LPS-induced IL-1β secretion in both dose- and time-dependent manners, although the transcriptional levels of TNFα and IL-1β were not significant potentiated. This dissociation between the transcriptional induction and release strongly suggests an involvement of a post-translational regulatory mechanism (Lopez-Castejon and Brough 2011). The IL-1β is released as a proprotein (pro-IL-1β) that needs to be proteolytically processed into its active form. In microglia, the processing of pro-IL-1β is mediated by the NLRP3 inflammasome at the post-translational level (Walsh et al. 2014). The inflammasome is a multiprotein complex, which serves as a caspase-1 activation platform. Active caspase-1 is the cleaving enzyme for processing pro-IL-1β into its mature, biologically active forms. Typically, two independent signals are required to fully activate the NLRP3 inflammasome (Hanamsagar et al. 2012). The first signal is to upregulate the major substrate (pro-IL-1β) and essential components (NLRP3) required for inflammasome activation. Because pro-IL-1β and NLRP3 are not constitutively expressed in microglia, additional factors are needed for transcriptional induction (Dostert et al. 2008; Halle et al. 2008). Although our goal is to investigate the role of the inflammasome-mediated proinflammatory signal in Meth-potentiation of HIV-associated neurotoxicity, little is known about HIV-1 protein as the first signal for the inflammasome activation. LPS is typically used as the first priming signal on microglia for inflammasome studies (Halle et al. 2008; Shi et al. 2012; Sarkar et al. 2017). The process of the LPS-primed NLRP3 inflammasome is well characterized and involved with the transcriptional induction (pro-IL-1β and NLRP3 induction) and multiple posttranslational events (extracellular signal regulated kinase 1 phosphorylation, proteasome inhibition, and NLRP3 deubiquitination) (Juliana et al. 2012; Ghonime et al. 2014). Based on these reasons, LPS was chosen as the stimulating factor in this study to mimic the priming stimulation of neuronal DAMPs or of the HIV proteins in the virus-bearing brain.
Currently, the role of Meth on inflammasome activation is not fully depicted. To investigate whether the inflammasome is the molecular target that responds to Meth stimuli, we systematically studied the inflammasome activation as a common downstream receptor locus for convergence of signals from multiple danger-associated pathways. The inflammasome consists of NLRP3 (sensor receptor), ASC (bridge protein), and caspase-1 (IL-1β converting enzyme) and can be activated in response to various upstream cellular danger-associated signals. As the primary mediator for pro-IL-1β maturation, NLRP3 inflammasome activation is always accompanied by processing of pro-IL-1β, cleavage of caspase-1 and ASC protein aggregation (Hanamsagar et al. 2012). After sequential stimulation with LPS and Meth, IL-1β and caspase-1 were cleaved to their activated forms, as illustrated by their lower molecular weights. The levels of aggregated ASC proteins were also elevated in response to co-application of LPS and Meth. These rsults are in an agreement with the classical cellular pattern of inflammasome activation, in which principal components redistribute from dispersed to clustered and further promote restoration of caspase-1 enzyme activity after the cross-cleavage process (Hanamsagar et al. 2012). As expected, the activity of caspase-1 was elevated along with increased concentration of Meth, and inhibition of caspase-1 totally reversed the enhancement of LPS-induced IL-1β processing and release. It worth noting that the Ac-YVAD-cmk also has some activity against caspase-4 and 5, which are all belong to the same family of inflammatory caspases with caspase-1 (the group I caspases). It has been well-recognized as a potent inflammasome inhibitor, and hence to exert anti-inflammatory and anti-pyroptotic effects (Van Opdenbosch et al. 2014). These results suggest a specific role of inflammasome activation in Meth potentiation of LPS-induced IL-1β processing and release.
Our results showed that Meth alone did not upregulate pro-IL-1β. This suggests that Meth may serve as a second signal for inflammasome activation. However, the target(s) for Meth-induced inflammasome activation is (are) still unclear. To address this question, the levorotatory form of Meth (L-Meth) was introduced. Meth is well-known to bind with plasma and vesicular monoamine transporters. In contrast, L-Meth has a much lower binding affinity with these monoamine transporters and thus is also less potent than the dextrorotatory form of Meth (Melega et al. 1999). Our ELISA results demonstrated that, without the LPS priming signal, none of the enantiomeric forms of Meth could induce the detectable IL-1β release. However, treatment of Meth (both D- and L- forms) after introduction of the priming signal LPS indeed significantly promoted the secretion of IL-1β in a dose-dependent manner. Thus, monoamine transporters are very unlikely to be the candidate receptors that mediate the potentiation effect of Meth. Due to the limited information on the differential binding affinity of the two enantiomers on other Meth-target receptors such as sigma-1 and trace amine-associated receptor 1, it is still difficult to draw a contour on the targets on which both Meth and LPS act.
Although it has been proven that the potentiation of Meth on LPS-induced IL-1β processing and release is mediated by the inflammasome, the mechanism is still unknown. Typically, it could be achieved by upregulation of the main components of the inflammasome or by the activation of upstream danger signaling pathways. However, our results showed that Meth had no significant effects on the levels of NLRP3 expression and the production of pro-caspase-1, indicating that Meth does not regulate the production of inflammasome components. As Meth has been reported to induce cell damage and distress, particularly in mitochondria- and lysosome-related pathways (Cubells et al. 1994; Potula et al. 2010; Nara et al. 2012) and the primary cellular targets for Meth in peripheral immune cells are mitochondrial ROS activation and lysosome disruption, which are well-accepted upstream pathways for inflammasome activation (Hanamsagar et al. 2012), we hypothesized that Meth treatment may trigger endogenous stress signals in microglia leading to Meth-induced inflammasome activation. To test the hypothesis, we used specific pharmacological inhibitors to dissect the Meth-associated danger signaling pathways. Application of the specific inhibitors targeting the mitochondrial ROS and lysosomal protease cathepsin B significantly blocked the Meth-induced inflammasome activation in a dose-dependent manner, demonstrating that Meth activates the inflammasome through the above two signaling pathways. However, it is worth pointing out that the blocking effect of the specific inhibitors may also be mediated by an off-target action. However, more experiments on these two subcellular pathways are need because RNA interference and cathepsin-B−/− cell lines failed to replicate CA-074Me effects in another study (Newman et al. 2009).
Further studies were performed with specific molecular probe and immunofluorescent staining to prove that the lysosomal disruption is specific to the effect of Meth potentiation. Compared to the low fluorescence levels in the control and the LPS alone groups, there was a dose-dependent increase in both total ROS and mitochondrial ROS signals after treatment with different concentrations of Meth. Because the mitochondrial ROS system also belongs to the total ROS system, the increased proportion of mitochondrial ROS was prallel with the change of total ROS, suggesting that the mitochondria is the primary source of Meth-induced ROS production. Previous studies indicate that incubation of microglia with a high concentration (1 μg/ml, 12 h) of LPS could induce mitochondrial ROS activation (Park et al. 2015). However, the concentration of LPS we used here is much lower (100 ng/ml, 6 h), and a washout step was carried out before addition of Meth. This may explain why we did not observe the elevation of mitochondrial ROS signal in the LPS treatment group.
Besides, we further evaluated the Meth-induced lysosomal membrane permeabilization (LMP) via lysosomotropic fluorochrome (LysoTracker) that accumulate inside acidic organelles. Both the reduced fluorescence intensity of LysoTracker and the diffuse distribution pattern of cathepsin B were detected in Meth-treated microglia, which are signs of lysosomal dysfunction. The decreased LysoTacker fluorescence is not entirely specific for LMP, which may also reflect an increase of lysosomal pH (Boya et al. 2003). However, the loss of pH gradient does not result in the translocation of cathepsin B. Therefore, our results of the reduction of LysoTracker fluorescence intensity were very likely an indicator of LMP but not merely the loss of pH gradient. LAMP-1 is a glycoprotein constituent of the lysosomal membrane that is constitutively expressed to resist hydrolase digestion (Eskelinen 2006). We did not observe a noticeable reduction of the LAMP-1 specific signal after Meth administration. The constant expression of LAMP-1 signal suggests that Meth does not completely collapse the lysosome, which is an inhibition signal for the inflammasome (Katsnelson et al. 2016). It needs to be emphorsized that the degree of lysosome impairment is also a key element for determining the ultimate result. Recent evidence indicates that while partial lysosome rupture may lead to inflammasome activation, complete lysosome rupture is a route to necrosis but not to the inflammasome (Guicciardi and Gores 2013). The uncoupling of cathepsin B and LAMP-1 signal indicates a low-level disruption of lysosomal membrane, but not an extensive rupture. Based on these reports, the lysosomal disruption may be another candidate mechanism for Meth-induced inflammasome activation.
Although our results indicate that both pathways mentioned above may play a role in Meth-induced inflammasome activation, it is still unknown whether Meth pretreatment directly activates these pathways. Some studies suggest there is a convergence point merged from multiple danger signaling pathways that directly activates the NLRP3 (Tschopp and Schroder 2010; Munoz-Planillo et al. 2013). However, recent evidence indicates a complicated interplay between mitochondria and lysosomes in initial NLRP3 activation (Heid et al. 2013) which is supported by another study showing the interaction between disruption of the mitochondria and the lysosome followed by NLRP3 inflammasome activation, suggesting that release of mitochondrial ROS might precede and cause lysosomal membrane permeabilization (Heid et al. 2013). Therefore, the possibility of lysosomal damage as the subsequent result of Meth-induced mitochondrial ROS cannot be entirely ruled out.
It is still not clear whether there are specific receptors expressed on lysosomes or mitochondria that directly interact with Meth at physiological concentrations. Increasing evidence demonstrates that the NLRP3 inflammasome assembles at the ER-mitochondrial interface (Misawa et al. 2013), suggesting a possible direct interaction of mitochondria and newly formed inflammasome. As lysosomal membrane permeability is also regulated by ROS production (Boya and Kroemer 2008), the Meth-induced inflammasome activation is likely governed by mitochondrial disruption, which is followed by lysosomal damage. Nevertheless, further studies are necessary to investigate whether a receptor for Meth presents on mitochondria while the lysosome functions as a downstream signal.
Finally, the effect of Meth potentiation of LPS-induced neurotoxicity is also examined. Previous results have demonstrated that LPS significantly activates the microglia in a dose-dependent manner and induces a potent neurotoxic effect (Houdek et al. 2014). In our in vitro neuroinflammation model, LPS was applied before Meth treatment to mimic pre-existing inflammatory stimulation. After priming with LPS, the supernatants of Meth-treated microglia significantly increased the percentage of apoptotic neurons compared to LPS alone. Without the priming signal of LPS, Meth also induced apoptosis of neurons at high concentration, but with much less potent effect. We found that inhibition of Meth-induced inflammasome activation by specific inhibitors reduced number apoptotic neurons, indicating that preventing inflammasome activation could be a potential strategy against Meth-induced neurotoxicity.
It is worth pointing out that this study was primarily focused on the Meth-induced inflammasome activation in microglia and its subsequent potentiation effect on neurotoxicity. Whether IL-1β was necessary or sufficient for microglial neurotoxicity induced by LPS and Meth remains to be determined. Currently, we have no direct evidence to conclude the Meth-potentiated neurotoxicity is solely mediated by the over-released mature form of IL-1β in supernatants. As IL-1β, on the one hand, has been shown to be neurotoxic by others (Ye et al. 2013; Brough et al 2015; Stojakovic et al. 2017), and the released active IL-1β in response to Meth-induced inflammasome activation, on the other hand, may also have a positive feedback on microglial inflammatory responses, which could promote the release of IL-1β and/or other neurotoxic factors and confound the neurotoxic effects observed in this study.
Taken together, our results provide a novel insight into Meth influence on the resident immune system within the CNS and a potential explanation of why Meth abusers suffer a more aggressive progression of many other neuroinflammatory-associated neurodegenerative disorders than non-users (Rippeth et al. 2004; Callaghan et al. 2012). Because LPS is a broad pro-inflammatory stimulus, the mechanisms underlying Meth potentiation on IL-1β production via inflammasome activation may have implications for the co-morbidity of other Meth-associated neurological conditions, such as Meth exacerbation of HIV-associated neurocognitive disorders. Since neuroinflammation itself contributes to many behavioral changes and cognitive impairment (Zerrate et al. 2007; Lee et al. 2008; Wager-Smith and Markou 2011), uncovering an association of Meth abuse and enhancement of pre-existing neuroinflammation could help prevent exacerbation of disease progression. Thus, therapeutic strategies targeting neuroinflammation could be a promising approach for Meth dependency, which is currently under investigation in a Phase 2 clinical trial (IBUD Ph II NCT01860807).
References
Attarzadeh-Yazdi G, Arezoomandan R, Haghparast A (2014) Minocycline, an antibiotic with inhibitory effect on microglial activation, attenuates the maintenance and reinstatement of methamphetamine-seeking behavior in rat. Prog Neuro-Psychopharmacol Biol Psychiatry 53:142–148
Beardsley PM, Shelton KL, Hendrick E, Johnson KW (2010) The glial cell modulator and phosphodiesterase inhibitor, AV411 (ibudilast), attenuates prime- and stress-induced methamphetamine relapse. Eur J Pharmacol 637:102–108
Boya P, Kroemer G (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27:6434–6451
Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, Perfettini JL, Kroemer G (2003) Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene 22:3927–3936
Brough D, Rothwell NJ, Allan SM (2015) Interleukin-1 as a pharmacological target in acute brain injury. Exp Physiol 100:1488–1494
Callaghan RC, Cunningham JK, Sykes J, Kish SJ (2012) Increased risk of Parkinson's disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend 120:35–40
Chang L, Alicata D, Ernst T, Volkow N (2007) Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction 102(Suppl 1):16–32
Colton CA (2009) Heterogeneity of microglial activation in the innate immune response in the brain. J NeuroImmune Pharmacol 4:399–418
Cubells JF, Rayport S, Rajendran G, Sulzer D (1994) Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci 14:2260–2271
Degenhardt L, Mathers B, Guarinieri M, Panda S, Phillips B, Strathdee SA, Tyndall M, Wiessing L, Wodak A, Howard J, Reference Group to the United Nations on HIV, injecting drug u (2010) Meth/amphetamine use and associated HIV: implications for global policy and public health. Int J Drug Policy 21:347–358
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677
Eskelinen EL (2006) Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Asp Med 27:495–502
Flora G, Lee YW, Nath A, Maragos W, Hennig B, Toborek M (2002) Methamphetamine-induced TNF-alpha gene expression and activation of AP-1 in discrete regions of mouse brain potential role of reactive oxygen intermediates and lipid peroxidation. Neuromolecular Med 2:71–85
Frank MG, Weber MD, Watkins LR, Maier SF (2015) Stress sounds the alarmin: the role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun 48:1–7
Frank MG, Adhikary S, Sobesky JL, Weber MD, Watkins LR, Maier SF (2016) The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav Immun 51:99–108
Friend DM, Keefe KA (2013) Glial reactivity in resistance to methamphetamine-induced neurotoxicity. J Neurochem 125:566–574
Funakoshi-Hirose I, Aki T, Unuma K, Funakoshi T, Noritake K, Uemura K (2013) Distinct effects of methamphetamine on autophagy-lysosome and ubiquitin-proteasome systems in HL-1 cultured mouse atrial cardiomyocytes. Toxicology 312:74–82
Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273:32608–32613
Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, Gavrilin MA, Wewers MD (2014) Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol 192:3881–3888
Glasner-Edwards S, Mooney LJ, Marinelli-Casey P, Hillhouse M, Ang A, Rawson R, Methamphetamine Treatment Project Corporate A (2010) Anxiety disorders among methamphetamine dependent adults: association with post-treatment functioning. Am J Addict 19:385–390
Gonzalez H, Elgueta D, Montoya A, Pacheco R (2014) Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J Neuroimmunol 274:1–13
Guicciardi ME, Gores GJ (2013) Complete lysosomal disruption: a route to necrosis, not to the inflammasome. Cell Cycle 12:1995–1995
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865
Hanamsagar R, Hanke ML, Kielian T (2012) Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol 33:333–342
Hashimoto K, Tsukada H, Nishiyama S, Fukumoto D, Kakiuchi T, Iyo M (2007) Protective effects of minocycline on the reduction of dopamine transporters in the striatum after administration of methamphetamine: a positron emission tomography study in conscious monkeys. Biol Psychiatry 61:577–581
Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD (2013) Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191:5230–5238
Hill PA, Buttle DJ, Jones SJ, Boyde A, Murata M, Reynolds JJ, Meikle MC (1994) Inhibition of bone resorption by selective inactivators of cysteine proteinases. J Cell Biochem 56:118–130
Houdek HM, Larson J, Watt JA, Rosenberger TA (2014) Bacterial lipopolysaccharide induces a dose-dependent activation of neuroglia and loss of basal forebrain cholinergic cells in the rat brain. Inflamm Cell Signal 1(1):e47
Jayanthi S, Deng X, Noailles PA, Ladenheim B, Cadet JL (2004) Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J 18:238–251
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287:36617–36622
Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA, Dubyak GR (2016) NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol 311:C83–C100
Kelly KA, Miller DB, Bowyer JF, O'Callaghan JP (2012) Chronic exposure to corticosterone enhances the neuroinflammatory and neurotoxic responses to methamphetamine. J Neurochem 122:995–1009
Krasnova IN, Cadet JL (2009) Methamphetamine toxicity and messengers of death. Brain Res Rev 60:379–407
LaVoie MJ, Card JP, Hastings TG (2004) Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol 187:47–57
Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT (2008) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 5:37
Liang HL, Arsenault J, Mortensen J, Park F, Johnson CP, Nilakantan V (2009) Partial attenuation of cytotoxicity and apoptosis by SOD1 in ischemic renal epithelial cells. Apoptosis 14:1176–1189
Liu L, Hutchinson MR, White JM, Somogyi AA, Coller JK (2009) Association of IL-1B genetic polymorphisms with an increased risk of opioid and alcohol dependence. Pharmacogenet Genomics 19:869–876
Liu L, Coller JK, Watkins LR, Somogyi AA, Hutchinson MR (2011) Naloxone-precipitated morphine withdrawal behavior and brain IL-1beta expression: comparison of different mouse strains. Brain Behav Immun 25:1223–1232
Liu J, Xu C, Chen L, Xu P, Xiong H (2012) Involvement of Kv1.3 and p38 MAPK signaling in HIV-1 glycoprotein 120-induced microglia neurotoxicity. Cell Death Dis 3:e254
Loftis JM, Janowsky A (2014) Neuroimmune basis of methamphetamine toxicity. Int Rev Neurobiol 118:165–197
Lopez-Castejon G, Brough D (2011) Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 22:189–195
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365
Mashayekhi V, Eskandari MR, Kobarfard F, Khajeamiri A, Hosseini MJ (2014) Induction of mitochondrial permeability transition (MPT) pore opening and ROS formation as a mechanism for methamphetamine-induced mitochondrial toxicity. Naunyn Schmiedeberg's Arch Pharmacol 387:47–58
Melega WP, Cho AK, Schmitz D, Kuczenski R, Segal DS (1999) L-methamphetamine pharmacokinetics and pharmacodynamics for assessment of in vivo deprenyl-derived l-methamphetamine. J Pharmacol Exp Ther 288:752–758
Melega WP, Cho AK, Harvey D, Lacan G (2007) Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse 61:216–220
Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14:454–460
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153
Nakajima A, Yamada K, Nagai T, Uchiyama T, Miyamoto Y, Mamiya T, He J, Nitta A, Mizuno M, Tran MH, Seto A, Yoshimura M, Kitaichi K, Hasegawa T, Saito K, Yamada Y, Seishima M, Sekikawa K, Kim HC, Nabeshima T (2004) Role of tumor necrosis factor-alpha in methamphetamine-induced drug dependence and neurotoxicity. J Neurosci 24:2212–2225
Nara A, Aki T, Funakoshi T, Unuma K, Uemura K (2012) Hyperstimulation of macropinocytosis leads to lysosomal dysfunction during exposure to methamphetamine in SH-SY5Y cells. Brain Res 1466:1–14
Newman ZL, Leppla SH, Moayeri M (2009) CA-074Me protection against anthrax lethal toxin. Infect Immun 77:4327–4336
Northrop NA, Yamamoto BK (2015) Methamphetamine effects on blood-brain barrier structure and function. Front Neurosci 9:69
Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, Kong IK, Chang KT, Lee DS (2015) Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci Lett 584:191–196
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nature reviews 6:193–201
Potula R, Hawkins BJ, Cenna JM, Fan S, Dykstra H, Ramirez SH, Morsey B, Brodie MR, Persidsky Y (2010) Methamphetamine causes mitrochondrial oxidative damage in human T lymphocytes leading to functional impairment. J Immunol 185:2867–2876
Prakash MD, Tangalakis K, Antonipillai J, Stojanovska L, Nurgali K, Apostolopoulos V (2017) Methamphetamine: effects on the brain, gut and immune system. Pharmacol Res 120:60–67
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462
Quinton MS, Yamamoto BK (2006) Causes and consequences of methamphetamine and MDMA toxicity. AAPS J 8:E337–E347
Rippeth JD, Heaton RK, Carey CL, Marcotte TD, Moore DJ, Gonzalez R, Wolfson T, Grant I (2004) Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc 10:1–14
Riviere GJ, Gentry WB, Owens SM (2000) Disposition of methamphetamine and its metabolite amphetamine in brain and other tissues in rats after intravenous administration. J Pharmacol Exp Ther 292:1042–1047
Robson MJ, Turner RC, Naser ZJ, McCurdy CR, O'Callaghan JP, Huber JD, Matsumoto RR (2014) SN79, a sigma receptor antagonist, attenuates methamphetamine-induced astrogliosis through a blockade of OSMR/gp130 signaling and STAT3 phosphorylation. Exp Neurol 254:180–189
Salamanca SA, Sorrentino EE, Nosanchuk JD, Martinez LR (2014) Impact of methamphetamine on infection and immunity. Front Neurosci 8:445
Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG (2017) Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson's disease. NPJ Parkinsons Dis 3:30
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675
Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH, Grant I (2007) Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev 17:275–297
Sekine Y, Iyo M, Ouchi Y, Matsunaga T, Tsukada H, Okada H, Yoshikawa E, Futatsubashi M, Takei N, Mori N (2001) Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am J Psychiatry 158:1206–1214
Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, Kawai M, Takebayashi K, Yamamoto S, Matsuzaki H, Ueki T, Mori N, Gold MS, Cadet JL (2008) Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci 28:5756–5761
Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, Yin X, Zhao D (2012) The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J Neuroinflammation 9:73
Shiba T, Yamato M, Kudo W, Watanabe T, Utsumi H, Yamada K (2011) In vivo imaging of mitochondrial function in methamphetamine-treated rats. NeuroImage 57:866–872
Snider SE, Hendrick ES, Beardsley PM (2013) Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol 701:124–130
Stojakovic A, Paz-Filho G, Arcos-Burgos M, Licinio J, Wong ML, Mastronardi CA (2017) Role of the IL-1 pathway in dopaminergic neurodegeneration and decreased voluntary movement. Mol Neurobiol 54:4486–4495
Thomas DM, Kuhn DM (2005a) MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res 1050:190–198
Thomas DM, Kuhn DM (2005b) Attenuated microglial activation mediates tolerance to the neurotoxic effects of methamphetamine. J Neurochem 92:790–797
Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM (2004) Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther 311:1–7
Tian C, Murrin LC, Zheng JC (2009) Mitochondrial fragmentation is involved in methamphetamine-induced cell death in rat hippocampal neural progenitor cells. PLoS One 4:e5546
Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10:210–215
Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M (2014) Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun 5:3209
Wager-Smith K, Markou A (2011) Depression: a repair response to stress-induced neuronal microdamage that can grade into a chronic neuroinflammatory condition? Neurosci Biobehav Rev 35:742–764
Walker NP, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett MC, Hammill LD et al (1994) Crystal structure of the cysteine protease interleukin-1 beta-converting enzyme: a (p20/p10)2 homodimer. Cell 78:343–352
Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97
Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF (2015) Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 35:316–324
Wu CW, Ping YH, Yen JC, Chang CY, Wang SF, Yeh CL, Chi CW, Lee HC (2007) Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol Appl Pharmacol 220:243–251
Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, Qian G, Zheng JC (2013) IL-1beta and TNF-alpha induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem 125:897–908
Zerrate MC, Pletnikov M, Connors SL, Vargas DL, Seidler FJ, Zimmerman AW, Slotkin TA, Pardo CA (2007) Neuroinflammation and behavioral abnormalities after neonatal terbutaline treatment in rats: implications for autism. J Pharmacol Exp Ther 322:16–22
Zhang L, Kitaichi K, Fujimoto Y, Nakayama H, Shimizu E, Iyo M, Hashimoto K (2006) Protective effects of minocycline on behavioral changes and neurotoxicity in mice after administration of methamphetamine. Prog Neuro-Psychopharmacol Biol Psychiatry 30:1381–1393
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225
Acknowledgements
We would like to thank Professor Myron L. Toews (Ph.D., University of Nebraska Medical Center; Department of Pharmacology and Experimental Neuroscience) for his critical reading the manuscript and valuable comments that significantly improved the manuscript. Special thanks extend to Professor T. Celeste Napier (Ph.D., Department of Psychiatry, Rush University, Chicago) for her valuable suggestions on this project. This work was supported by by NIH grant R01 NS077873.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of University of Nebraska Medical Center, Omaha, NE, USA.
Competing Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Xu, E., Liu, J., Liu, H. et al. Inflammasome Activation by Methamphetamine Potentiates Lipopolysaccharide Stimulation of IL-1β Production in Microglia. J Neuroimmune Pharmacol 13, 237–253 (2018). https://doi.org/10.1007/s11481-018-9780-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-018-9780-y