
Abstract
The heavy chain subunit of ferritin (FHC), a ubiquitous protein best known for its iron-sequestering activity as part of the ferritin complex, has recently been described as a novel inhibitor of signaling through the chemokine receptor CXCR4. Levels of FHC as well as its effects on CXCR4 activation increase in cortical neurons exposed to mu-opioid receptor agonists such as morphine, an effect likely specific to neurons. Major actions of CXCR4 signaling in the mature brain include a promotion of neurogenesis, activation of pro-survival signals, and modulation of excitotoxic pathways; thus, FHC up-regulation may contribute to the neuronal dysfunction often associated with opiate drug abuse. This review summarizes our knowledge of neuronal CXCR4 function, its regulation by opiates and the role of FHC in this process, and known mechanisms controlling FHC production. We speculate on the mechanism involved in FHC regulation by opiates and offer FHC as a new target in opioid-induced neuropathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytokines are a family of small, secreted proteins well known for their ability to regulate host responses to infection, inflammation, and trauma. Among these, the class of chemotactic cytokines or chemokines specializes in attracting receptor-bearing target cells (Charo and Ransohoff 2006). The functions of chemokine-induced cellular migration vary depending on the specific ligands and receptors involved as well as tissue type and stage of development, although two broad types of effects can be described: classically, inducible chemokine production facilitates inflammatory processes through the recruitment of leukocytes to local sites of infection, and, in a more recently appreciated manner, constitutive chemokine production supports homeostatic tissue maintenance and developmental processes. Although most studies have focused on the roles of chemokines in inflammatory conditions, growing evidence supports important non-inflammatory functions, including the regulation of homeostatic lymphocyte trafficking, hematopoiesis, and cellular guidance during tissue development. In certain conditions, chemokines may also contribute to cancer cell proliferation, migration, and invasion.
Particularly interesting among the regulatory functions of chemokines are those occurring in the brain, where chemokines are constitutively expressed in both neurons and glia (Lavi et al. 1998; Callewaere et al. 2007). Evidence that chemokines can modify diverse brain properties, including synaptic activity, axonal guidance, adult neurogenesis, neuronal survival and migration, and microglia activation supports an important modulatory role of these proteins at all levels of brain function. Much of this research has focused on the chemokine CXCL12 and its major (if not exclusive) signaling receptor CXCR4, due to their constitutive expression and involvement in essential functional processes.
Interestingly, in a theme first demonstrated in immune-modulating chemokines (Rogers and Peterson 2003; Szabo et al. 2003), signaling of chemokines in the brain has been shown to undergo attenuation following treatment with opioids (Sengupta et al. 2009), a process that may contribute to neuronal dysfunction associated with opiate drug abuse (Nath 2010). Mu-opiate-induced neuronal CXCR4 inhibition has recently been shown to rely on an increased association of the receptor with ferritin heavy chain (FHC), an unexpected protein regulator of CXCR4 signaling (Li et al. 2006; Sengupta et al. 2009). Considering the importance of the signaling pathways modulated by CXCR4 activity, this novel interaction likely impairs a variety of healthy neuronal homeostatic processes. An emerging theme of current research is that of better characterizing the regulatory mechanisms underlying opiate effects on FHC, as well as exploring the impact of increased FHC levels on homeostatic neuronal functions. This review will discuss the role of FHC in mediating effects of opiates on CXCR4 signaling, mainly in neurons, and will provide speculation as to the mechanisms underlying opiate-induced changes in neuronal FHC levels.
Homeostatic roles of CXCL12 in the brain
Both CXCL12 and its major signaling receptor CXCR4 are constitutively expressed in the developing and adult brain, suggesting a fundamental role of this signaling pair in brain function. Indeed, knockout mice for either CXCL12 or CXCR4 exhibit severe abnormalities in brain development and die prenatally (Ma et al. 1998; Zou et al. 1998; Bagri et al. 2002; Lu et al. 2002). Here, we will only briefly review the major regulatory roles of CXCL12 in the brain, as others have discussed this topic more comprehensively (Tran and Miller 2003; Li and Ransohoff 2008).
In the developing brain, CXCL12 promotes the proliferation, survival, and proper migration of neuronal precursor cells (Li and Ransohoff 2008). Embryonic CXCR4 expression in the neurogenic ventricular, subventricular, and marginal zones is vital for the production of neuronal precursor cells. Complex neuronal migration patterns, particularly those of cortical Cajal–Retzius cells, cerebellar granule precursor cells, and dentate gyrus granule precursor cells also rely on CXCR4 signaling (Li and Ransohoff 2008). Subsequent establishment of precise synaptic connections, a process depending on attractive and repulsive axonal guidance cues, is also modulated by CXCR4 signaling (Chalasani et al. 2003; Li et al. 2005; Chalasani et al. 2007). CXCL12 similarly promotes neural precursor cell proliferation and migration in adult brain, (Imitola et al. 2004; Dziembowska et al. 2005; Robin et al. 2006), a process that may contribute to tissue repair or functional adaptation.
In the mature brain, actions of CXCL12 involve a variety of mechanisms directly and indirectly promoting neuronal survival and neurotransmission. CXCL12 activates the anti-apoptotic kinase Akt (Khan et al. 2004), effectively inhibiting the apoptotic cascade in cultured neurons. It also promotes differentiation of oligodendrocyte precursors and induces remyelination in animal models (Patel et al. 2010). CXCL12 enhances activity of the transcriptional repressor protein Rb, whose constitutive repression of aberrant cell cycle proteins in post-mitotic neurons prevents entry into apoptosis (Khan et al. 2003). Notably, Rb expression was required for the neuroprotective activity of CXCL12, as demonstrated in the context of excitotoxic insults (Khan et al. 2008). CXCL12 has also been shown to increase production of soluble fractalkine or CX3CL1 (Cook et al. 2010), a chemokine capable of preventing neurotoxicity associated with microglial activation (Cardona et al. 2006). Another line of investigation has explored effects of CXCL12 on synaptic transmission. In this context, CXCL12 was shown to regulate subunit composition of the NMDA-type glutamate receptor both in vitro and in vivo, altering NMDA-induced calcium responses associated with excitotoxicity. Interestingly, this pathway selectively decreased calcium responses evoked following activation of extrasynaptic NMDA receptors (often a pathological event), while preserving pro-survival pathways elicited by synaptic NMDA receptor activation (Nicolai et al. 2010). Electrophysiologically, these findings are supported by the ability of CXCL12 to induce synaptic depression, through a direct modulation of voltage-dependent currents and a decrease in evoked glutamate release (Ragozzino et al. 2002; Guyon et al. 2005).
The role of the CXCL12/CXCR4 axis in neuronal survival was also studied in the human brain under pathological conditions, specifically HIV infection (Zhang et al. 2003; Vergote et al. 2006). These authors demonstrated that proteolytic cleavage of CXCL12 impairs its ability to stimulate CXCR4 and leads to neurotoxicity via interaction of cleaved CXCL12 with a different receptor (CXCR3). Importantly, proteolytic processing of CXCL12 is increased in HIV-positive subjects, particularly those with HIV-associated dementia. Thus, stimulation of the CXCL12/CXCR4 axis normally exerts a neuroprotective action through its effects on different cell types, including neurons. Although mechanistically diverse, these various modulatory actions of CXCL12 in the brain contribute to a powerful pro-survival signal, the disruption of which may severely affect healthy brain function.
Regulation of CXCR4 function by opiates
An emergent concept in the study of opioid physiology concerns a distinct interrelatedness with the cytokine family, a principle known as the “opioid–cytokine connection.” Opioids and cytokines share similar properties, such as release from immune cells, autocrine, paracrine, or endocrine signaling mechanisms, and elicitation of a wide variety of effects in different cell types. Moreover, both opioids and cytokines reciprocally modulate each other's signaling activity (Peterson et al. 1998) and may interact to form functionally distinct heterodimers (Chen et al. 2004; Pello et al. 2008). Chemokines in the brain are no exception to this phenomenon, and a recent series of experiments has identified an inhibitory role of opioid agonists in neuronal CXCR4 signaling (Patel et al. 2006; Sengupta et al. 2009).
Multiple mechanisms have been identified contributing to an opioid-induced decrease in CXCR4 activity. Simultaneous activation of delta opioid receptors (DOR) and CXCR4 leads to the formation of non-functional DOR/CXCR4 heterodimers incapable of responding to agonists of either receptor (Pello et al. 2008). Although first demonstrated in immune cells, this phenomenon likely occurs in glia as well, providing an interactive regulatory process between the opioid and chemokine systems (Burbassi et al. 2010). Kappa opioid receptors (KOR) have also been shown to undergo cross-desensitization with CXCR4 in immune cells; in this case, opioid-induced CXCR4 inhibition is achieved at least in part by receptor internalization, effectively decreasing levels of surface CXCR4 available to agonist (Finley et al. 2008). Activation of the third major class of opioid receptors, mu (MOR), additionally inhibits CXCR4 activity and has been explored intensively by our laboratory in recent years.
The MOR agonist DAMGO was first shown to decrease subsequent CXCR4 activation by CXCL12 in cortical neurons, as measured by downstream activation of the intracellular mediators ERK and Akt (Patel et al. 2006). This effect was observed in cultured neurons (in the presence or absence of glia) and occurred without apparent changes in surface or total levels of CXCR4. Similar effects were seen following treatment with the endogenous MOR agonist endomorphin-1 and with morphine, a powerful clinical analgesic and an active metabolite of heroin, suggesting a relevance of this pathway in therapeutic as well as illicit opiate use. Importantly, MOR agonism was also shown to block the neuroprotective actions of CXCL12 associated with NMDA-induced neurotoxicity, indicating a significant loss of CXCR4 function mediated by this pathway (Patel et al. 2006). The inhibitory effect of morphine on CXCR4 activation was also demonstrated in vivo, as measured by a decrease in G-protein coupling following CXCL12 treatment of brain slices from morphine-treated animals (Sengupta et al. 2009). Mechanistically, morphine was shown to prevent CXCL12-induced phosphorylation of CXCR4 at serine 338 (corresponding to serine 339 in humans), a site in the C terminus tail involved in signaling (Busillo et al. 2010) and to inhibit a CXCL12-induced reduction in surface CXCR4 (Sengupta et al. 2009). Interestingly, the ability of MOR agonism to inhibit subsequent CXCR4 signaling required prolonged agonist exposure, i.e., at least 6 h, and de novo protein synthesis (Sengupta et al. 2009), suggesting the involvement of a protein intermediate. Experiments conducted in glia cultures enriched in astrocytes showed that opiates do not alter CXCR4-mediated signaling in glia via similar mechanisms (Sengupta et al. 2009; Burbassi et al. 2010), though this does not exclude traditional mechanisms of heterologous desensitization as previously reported in various cells (Rogers et al. 2000; Devi 2001; Burbassi et al. 2010). Together, these data support a model in which prolonged MOR stimulation in the brain inhibits neuronal CXCR4 activation by its natural ligand, which promotes neurotoxicity in vulnerable cells (such as those exposed to toxic concentrations of glutamate).
Although different opioid receptor subtypes contribute in different ways to the inhibitory effects of opioids on CXCR4 signaling, the role of MOR may be particularly relevant to human pathology, given the frequent clinical use of MOR agonists as well as their widespread abuse. This pathway may play a major role in the neuronal dysfunction associated with continued opiate use and was recently discovered to involve actions of the novel CXCR4-interacting protein, FHC, as further explained below.
Ferritin heavy chain as a regulator of CXCR4 function
Ferritin is a widely expressed protein, best known for its ability to oxidize and sequester free intracellular iron in its less toxic ferric form. This function involves the coordinated activity of Heavy and Light Chain subunits (FHC and FLC, respectively), which come together as a 24 subunit complex in varying but cell-specific proportions to form an iron-sequestering shell (Hintze and Theil 2006). Ferroxidase activity, the oxidation of Fe2+ to the less reactive Fe3+, is inherent in FHC but not FLC, although FLC is important in nucleation of the iron core. These two subunits are normally co-expressed in a large variety of cells, and little is known about subunit-specific functions or interactions with other proteins.
The first evidence of a novel function of FHC, which may act independently from iron, came from a screen designed to identify novel binding partners of CXCR4, in which FHC was found to associate with both N and C termini of the receptor in human embryonic kidney (HEK293) cells (Li et al. 2006). This interaction was shown to increase following CXCL12 treatment, peaking after roughly 30 min. A regulatory function of the association was suggested by FHC overexpression studies, in which CXCL12-induced ERK activation and chemotaxis were significantly attenuated (Li et al. 2006). Conversely, FHC knockdown caused a prolonged activation of ERK and decreased CXCR4 internalization following CXCL12 treatment, suggesting a tonic inhibitory role in CXCR4 signaling.
Although the mechanism underlying the ability of this novel CXCR4 binding protein to regulate receptor signaling is not clear, the CXCL12-induced interaction of the two proteins was found to depend on phosphorylation of FHC, an event also required to functionally inhibit CXCR4 signaling (Li et al. 2006). Additionally, the CXCL12-induced FHC–CXCR4 interaction was enhanced by CXCR4 internalization in HEK293 cells (Li et al. 2006), suggesting a model in which FHC may inhibit CXCR4 recycling or return to an active conformation.
In light of these new findings linking FHC to alterations in CXCR4 signaling, we questioned whether FHC was involved in the effects of morphine on neuronal CXCR4. Initial findings demonstrated a mu-opioid-induced increase in FHC protein levels and CXCR4 association, both in vitro and in vivo, temporally correlating with the effects on CXCR4 signaling (Sengupta et al. 2009). Interestingly, prolonged stimulation of MOR by DAMGO did not increase FHC protein levels in cultured glia cells, reflecting our observed functional cell type specificity (i.e., as mentioned earlier, our previous studies showed that CXCR4 stimulation of ERK was unaffected under similar conditions (Sengupta et al. 2009)). In order to evaluate the relevance of FHC to CXCR4 inhibition by MOR agonists, FHC knockdown experiments were performed, demonstrating a diminished effect of mu-opioids on CXCL12-induced signaling. Importantly, these effects were confirmed in vivo, as FHC knockdown decreased the effects of MOR agonism on CXCL12-induced G-protein activation.
A relevance of these findings for human disease is supported by preliminary studies in post mortem human brain tissue, showing an elevation in numbers of FHC-positive cells in the frontal cortex of opiate drug abusers (Pitcher et al. 2010). An increase in cortical FHC was also observed in patients with HIV-associated dementia (Pitcher et al. 2010), a neurodegenerative complication of HIV with increased prevalence and rate of progression among opiate users (Davies et al. 1998; Bell et al. 2006; Hauser et al. 2006; Nath 2010). Importantly, this disease-associated elevation of FHC coincided with a decrease in phosphorylated (active) CXCR4, supporting a role of FHC-induced CXCR4 inhibition in pathological neuronal dysfunction (Pitcher et al. 2010).
Together, these studies identify a novel role of FHC in the regulation of CXCR4 signaling and suggest a new target in opiate-induced neuronal dysfunction. Although the mechanism and full functional consequences of neuronal FHC up-regulation are unclear, the diversity and importance of pathways likely inhibited by this effect suggest a broad physiological and pathological relevance and warrant further investigation into both its causes and effects. An aim of ongoing experiments in our laboratory is to characterize the mechanistic pathway initiated by MOR activation leading to enhanced FHC levels and understand the reasons for the observed differences in CXCR4 modulation by opiates between neurons and astroglia; an understanding of this process may suggest targets for intervention should regulating neuronal FHC prove therapeutically useful, and may broaden our knowledge of factors contributing to FHC regulation in health and disease.
FHC transcriptional regulation
Levels of cellular ferritin are dynamically controlled at both transcriptional and translational levels by a variety of factors involved in free iron and oxidative stress responses (Torti and Torti 2002). Although some genetic players vary between species, broad regulatory themes are conserved, highlighting the importance of ferritin to known iron-binding functions. Major transcriptional and translational regulatory mechanisms, mostly characterized in non-neuronal cells, are described below and illustrated in Fig. 1.

Transcriptional and translational control of FHC production: a depiction of the human FHC promoter region, containing the basal enhancer (B box), Sp1 binding site (A box), an inhibitory region (G-fer), antioxidant response element (ARE), and TSA responsive element (a). Processes illustrated in b regulate translation of FHC mRNA. Iron regulatory proteins (IRP) 1 and 2 bind the stem-loop structure of the iron responsive element (IRE) to inhibit translation and are inactivated by high levels of free iron. Mechanisms of iron-mediated IRP inactivation include a loss of IRE binding affinity (IRP1) and protein degradation (IRP2)
Basal FHC expression in humans is regulated by promoter activity at the so-called B box, a region of DNA 62 base pairs from the transcription start site (Fig. 1a). This region binds a protein complex known as the B box binding factor (Bbf), composed of the transcription factor NFY, the co-activator p300, and the histone acetylase p300/CBP associated factor (PCAF) (Bevilacqua et al. 1995; Faniello et al. 1999). Various intracellular signaling mediators modulate FHC transcription by promoting or inhibiting Bbf formation; transcriptional activity at this site is activated by cAMP (Bevilacqua et al. 1997) and c-Jun (Faniello et al. 2002), and is inhibited by the adenoviral oncogene E1A (Bevilacqua et al. 1997) and the tumor suppressor protein p53 (Faniello et al. 2008). Additionally, increasing amounts of PCAF may contribute to tissue-specific or developmental differences in FHC expression (Bevilacqua et al. 1998). A negative cis-acting element 291 base pairs from the transcription start site was discovered and named G-fer, for its long stretch of Gs (Barresi et al. 1994). This site likely binds inhibitory factor 1 (IF-1), a transcription factor often associated with G-rich promoter elements, enabling a repression of B box activity. A third active site identified within the human FHC promoter is the A box, 132 base pairs from the transcription start site, containing a consensus sequence for the transcription factor Sp1, which may promote transcriptional activity (Bevilacqua et al. 1992).
Transcriptional induction of FHC is most notably stimulated by inflammatory cytokines, presumably in an effort by the cell to limit ferrous iron's contribution to free radical generation and oxidative stress (Miller et al. 1991). Although observed in both humans and small animals, this molecular mechanism has been best characterized in the mouse. Many of these studies focused on TNF, a central mediator of the inflammatory response, which was shown to selectively induce FHC mRNA and protein levels in a variety of mesenchymal cell lines (Torti et al. 1988; Miller et al. 1991), although a similar effect was also induced by the inflammatory cytokine IL-1 (Wei et al. 1990). The promoter region involved in this response was identified in the mouse as separate from that interacting with Bbf-like factors, instead containing two NF-κB binding sites essential for transcriptional induction (Kwak et al. 1995). Although a corresponding regulatory element mediating this effect has not been described in the human FHC promoter, the molecular processes regulating basal FHC transcription in the mouse are similar to those in humans, with the exception of being located roughly 5 kb further 5′ (Tsuji et al. 1999).
A similar stimulation of FHC transcription has been demonstrated following pro-oxidant treatment; this effect relies on an antioxidant response element (ARE) or electrophile response element, common in promoters of genes regulating the response to oxidative stress (Tsuji et al. 2000). The human ARE is found 4.5 kb from the transcriptional start site, far upstream from other regulatory elements (Tsuji 2005). This region contains two essential AP1-like motifs involved in enhancer activity. Binding of the transcription factor Nrf2 likely promotes basal FHC expression, while oxidant treatment induces binding of JunD, a regulatory protein involved in antioxidant defense, mediating an increase in FHC transcription (Tsuji 2005).
A recent study (Wang et al. 2010a) identified inhibitors of histone deacetylases (HDACs), enzymes named for their ability to deacetylate histones and thereby inhibit local transcription, as activators of FHC expression. The region responsible for this effect is located 79 base pairs from the transcriptional start site, and was named the TSA responsive element. Unlike typical HDACs, however, in this context, HDAC inhibitors likely function by increasing recruitment of the transcription factor NFY to the promoter independently of histone acetylation, possibly by acetylating NFY. The physiological relevance of this finding is unknown.
Together, these studies demonstrate a well-conserved basal transcription apparatus in the FHC promoter, capable of responding to inflammatory mediators or oxidative stress by increasing gene expression. Although transcriptional effects likely evolved to control the cell's oxidative processes, these molecular mechanisms may also influence CXCR4 activity or undergo alterations in response to prolonged opiate exposure, thus mediating pathological FHC dysregulation.
FHC translation and beyond
In addition to processes regulating FHC transcription, several mechanisms altering mRNA stability or rate of translation have been described, contributing to the dynamic control of FHC protein levels. Unlike transcriptional regulatory processes, these mechanisms respond primarily to levels of free intracellular iron (Theil 2007).
Rates of ferritin (i.e., FHC and FLC) translation are controlled in large part by a stem-loop structure in the 5′ untranslated region of the transcript termed the iron responsive element (IRE), present in several genes regulating iron homeostasis. The IRE can bind iron regulatory proteins 1 and 2 (IRP1 and IRP2), both of which inhibit ferritin translation (Torti and Torti 2002), as represented schematically in Fig. 1b. Levels of free iron regulate activity of both IRPs, although this is achieved through different mechanisms. In conditions of iron abundance, IRP1 exists as a cytosolic aconitase independent from the IRE, but decreasing iron levels promote formation of an iron–sulfur cluster causing the protein to bind the IRE and inhibit translation (Haile et al. 1992). IRP2, however, is regulated by protein abundance; IRP2 protein is expressed and readily binds the IRE when iron is scarce, but is rapidly degraded in the presence of iron (Guo et al. 1994, 1995).
Although several related proteins contain an IRE, including FLC, recent studies demonstrate differential activation of the FHC and FLC IREs in certain conditions, suggesting some control of the FHC:FLC ratio at the level of translation (Sammarco et al. 2008). Additionally, IRP1 and IRP2 may have distinct tissue-specific roles, as relative proportions of these proteins vary in different cell types, with a notable dominance of IRP1 over IRP2 levels in the brain (Thomson et al. 1999). Brain IRP2 is not entirely redundant or expendable, however, as IRP2 knockout mice exhibit a severe dysregulation of CNS iron levels and undergo neurodegeneration associated with accumulated ferritin and ferric iron (LaVaute et al. 2001).
Although IRP activity is best characterized in response to free intracellular iron levels, instances of iron-independent IRP regulation have also been described, suggesting an integration of signals occurring at the level of the IRPs. Interestingly, phosphorylation of IRP1 and particularly IRP2 by protein kinase C was shown to stimulate IRE binding, suggesting a possible mechanism whereby signal transduction pathways could modify rates of ferritin translation (Schalinske and Eisenstein 1996). Alterations in IRP2 phosphorylation states may also regulate cell-cycle-associated changes in ferritin synthesis in dividing cells (Wallander et al. 2008). Hypoxic conditions may modify IRP binding activities as well, although the data describing this effect are inconsistent; hypoxia was shown to increase IRP1 binding in human hepatoma, HEK293, and erythroleukemia cells (Toth et al. 1999; Christova and Templeton 2007), but to decrease its binding in rat hepatoma cells (Hanson and Leibold 1998) and mouse macrophages (Kuriyama-Matsumura et al. 2001). Furthermore, some mechanisms regulating IRP activity may overlap with those involved in gene transcription; oxidative stress has been shown to inactivate IRP1 (Cairo et al. 1996; Tsuji et al. 2000; Zumbrennen et al. 2009), and the inflammatory mediator NO+, released from activated macrophages, contributes to the degradation of IRP2 and production of ferritin (Mikhael et al. 2006). These oxidative and inflammatory effects likely contribute to the cytoprotective iron-limiting response. Interestingly, some evidence has indicated an increase in reactive oxygen species following prolonged morphine treatment both in vitro (Koch et al. 2009; Turchan-Cholewo et al. 2009) and in vivo (Pereska et al. 2007; Lam et al. 2008), which may contribute to the increase in FHC levels—although this remains to be established.
Additional post-transcriptional mechanisms regulating FHC synthesis involve modifying mRNA stability. An increase in T cell intracellular calcium levels was shown to increase FHC synthesis by increasing the mRNA half-life (MacKenzie and Tsuji 2008). Although the mechanism underlying mRNA stabilization and the relevance of this process in other cell types is unclear, this effect was thought to protect cells against reactive oxygen species and apoptotic signaling associated with calcium-induced T cell activation. Conversely, increased degradation or translational inhibition of FHC mRNA may be induced by specific microRNAs, thereby repressing protein synthesis. A particular role of miR-200b in repressing FHC translation was recently described in human breast cancer cells (Shpyleva et al. 2010), an effect which may play a role in regulating tumor-cell aggressiveness. This miRNA has also been shown to protect neurons from cell death associated with ischemia (Lee et al. 2010), making this an intriguing target linking FHC repression to neuronal survival.
Another notable mechanism regulating cellular ferritin levels involves the transferrin receptor (TfR1), recently described as a cell-surface receptor mediating the endocytosis of human FHC (Li et al. 2010). Several cell types have been shown to secrete ferritin, a process that is increased by iron as well as inflammatory cytokines, including IL-1 and TNFα (Wang et al. 2010b). Nonspecific inflammation is also associated with elevated serum ferritin, likely as a mechanism for cells to reduce intracellular iron levels, which may contribute to oxidative stress (Wang et al. 2010b). Although little is known concerning the cell's regulation of ferritin import, modification of this process may substantially alter intracellular protein levels.
By modulating the activities of key regulatory proteins, diverse stimuli combine to fine-tune ferritin protein levels within a cell. The existence of multiple independent regulatory pathways highlights the importance of a dynamic functional response and offers clues into the mechanism of a newly identified FHC regulator, morphine.
FHC: a new opiate target?
Together, these studies describe a novel role of FHC in neuronal function and survival and suggest a pathological impact of FHC dysregulation by morphine or other MOR agonists. Although the full extent of functional consequences associated with elevated FHC levels remains to be seen, processes regulating the expression or synthesis of the protein should be explored as potential therapeutic targets. Similarly, FHC may be considered as a downstream effector of changes induced by known FHC-producing stimuli, such as oxidative stress. The effect of oxidative stress on CXCR4 function, however, remains to be investigated.
An interesting feature of FHC regulation by morphine is its apparent specificity in the brain for neuronal cells. Although an inhibitory association of FHC with CXCR4 was first described in HEK293, HeLa, and Jurkat T cells (Li et al. 2006), the induction of FHC by morphine has only been examined in the brain, where the effect seems to be unique to neurons (Sengupta et al. 2009; Burbassi et al. 2010). Thus, FHC likely regulates CXCR4 activation in a wide variety of cell types, but the amplification of this effect by morphine may be specific to neurons. Major consequences of morphine abuse include inhibition of neuronal plasticity (Weber et al. 2006), neurodevelopmental defects, (Sargeant et al. 2008), changes in synaptic transmission and levels of NMDA receptor subunits (Ko et al. 2008; Johansson et al. 2010), and increased neurotoxicity in vulnerable cells (Gurwell et al. 2001; Lin et al. 2004), processes all of which are antagonized by neuronal CXCR4 activation and therefore may be modified by FHC. Regulation of FHC by morphine in additional cell types outside of the brain would extend the relevance of this finding to other cellular processes, whereas a strictly neuronal specificity would make FHC an attractive target for modulating the pro-survival effects of CXCR4 in the context of neurodegeneration. A neuronal specificity of this effect may also indicate a unique regulatory mechanism, distinct from the transcriptional and translational processes characterized in other cell types.
Another important consideration of this work is additional targets of elevated neuronal FHC beyond CXCR4. Although MOR agonism and concomitant FHC induction was unable to block survival pathways stimulated by another chemokine (CX3CL1) or the neurotrophic factor BDNF (Sengupta et al. 2009), FHC overexpression in HEK293 cells was shown to inhibit signaling downstream of the chemokine receptor CXCR2 by its natural ligand CXCL8 (Li et al. 2006), suggesting that additional receptors or signaling pathways may be modulated by FHC depending on cell type or other factors. Indeed, a broad role of FHC in the suppression of hematopoietic cell proliferation and differentiation (Recalcati et al. 2008), as well as an ability to induce apoptosis in primary hepatocytes in an autocrine manner (Bresgen et al. 2007), supports important iron-independent functions of this protein. FHC was furthermore shown to directly modify expression of other proteins; FHC positively regulates translation of an enzyme involved in folate metabolism through an interaction with mRNA-binding proteins (Woeller et al. 2007) and increases protein levels and transcriptional activity of the tumor suppressor p53 during increased oxidative stress (Lee et al. 2009). In addition to these novel iron-independent functions of FHC, alterations in iron homeostasis induced by FHC may also contribute to neuronal dysfunction associated with prolonged opiate use.
Another important distinction raised by these studies concerns the functional uniqueness of FHC apart from FLC, whose contribution is required to efficiently sequester iron. Although an induction of neuronal FLC in response to morphine has not yet been excluded, the association of FHC with CXCR4 appears to occur independently of FLC (Li et al. 2006), suggesting unique functional and regulatory properties of the heavy chain subunit. Mechanisms differentially regulating the two subunits generally operate at the level of transcription, including a specific induction of FHC in response to inflammation (Kwak et al. 1995) and oxidative stress (Tsuji 2005), although translational control by IRPs can also act subunit-specifically under certain circumstances (Sammarco et al. 2008). Another regulatory mechanism specific for the heavy chain subunit is the uptake of extracellular FHC mediated by the transferrin receptor (Li et al. 2010). In our studies, a morphine-induced increase in FHC levels was observed in a virtually pure neuronal culture (Sengupta et al. 2009), requiring increased neuronal production of the protein. However, little is known about the regulation of FHC import or export, which may modulate its functional capabilities in certain conditions as well.
Additional evidence supporting an important neurophysiological function of FHC comes from an apparent involvement of the protein in triple A syndrome, a poorly characterized neurological disorder. A majority of triple A syndrome patients exhibit mutations in the AAAS gene, which encodes a protein component of the nuclear pore complex recently shown to interact with FHC (Storr et al. 2009). Clinical features of the disorder are likely caused by impaired nuclear FHC uptake and decreased nuclear FHC content (Storr et al. 2009). Although the specific functions of nuclear FHC are unclear, nuclear translocation of the protein is induced following CXCL12 treatment (Li et al. 2006), suggesting a relevance of this process in the regulation of CXCR4 signaling. Furthermore, the specificity of this disease-associated transport deficit for the heavy chain subunit of ferritin and the associated neurological symptoms support a unique and important role of this protein in healthy neuronal function.
Although the mechanism linking MOR stimulation to increased neuronal FHC levels remains unclear, preliminary studies in our laboratory indicate that FHC mRNA levels are not altered (unpublished data), suggesting a post-transcriptional regulatory effect. Known mechanisms that would account for this observation involve translational inhibition mediated by changes in IRP activity or by specific miRNAs. Additionally, changes in the protein degradation rate may also be considered. The apparent specificity of this effect for FHC has led us to focus on translational regulation by miRNAs, which may underlie broad changes in neuronal function induced by morphine.
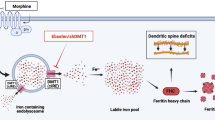
A better understanding of the regulatory processes contributing distinctly to FHC expression will help us evaluate the causes and effects of elevated FHC levels in the brain and elsewhere. As a modifier of neuronal CXCR4 function, FHC may alter a variety of homeostatic neuronal processes, many of which are known to be affected by chronic opiate use. Future studies, therefore, should aim to broaden our knowledge of physiological and pathological regulatory mechanisms specific for neuronal FHC and to more completely assess the role of elevated FHC in opiate-induced neuronal dysfunction. Figure 2 reports a simplified model depicting our current research efforts in this direction, focusing on the role of miR-200b in regulating FHC translation, and the contribution of transferrin-mediated endocytosis to FHC content and CXCR4 activation.

Additional mechanisms regulating FHC levels: in addition to transcriptional and IRP-mediated translational mechanisms, FHC levels can be regulated by specific miRNAs and by TfR1-mediated endocytosis. FHC mRNA is a known target of miR-200b and the effects of morphine on miR-200b production are currently under investigation in our laboratory. Extracellular FHC, produced in part by non-neuronal cells, may be an important source of the protein under certain conditions, such as chronic inflammation; the regulation of FHC endocytosis is another current focus of our laboratory
References
Bagri A, Gurney T, He X, Zou YR, Littman DR, Tessier-Lavigne M, Pleasure SJ (2002) The chemokine SDF1 regulates migration of dentate granule cells. Development 129:4249–4260
Barresi R, Sirito M, Karsenty G, Ravazzolo R (1994) A negative cis-acting G-fer element participates in the regulation of expression of the human H-ferritin-encoding gene (FERH). Gene 140:195–201
Bell JE, Arango JC, Anthony IC (2006) Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol 1:182–191
Bevilacqua MA, Giordano M, D'Agostino P, Santoro C, Cimino F, Costanzo F (1992) Promoter for the human ferritin heavy chain-encoding gene (FERH): structural and functional characterization. Gene 111:255–260
Bevilacqua MA, Faniello MC, D'Agostino P, Quaresima B, Tiano MT, Pignata S, Russo T, Cimino F, Costanzo F (1995) Transcriptional activation of the H-ferritin gene in differentiated Caco-2 cells parallels a change in the activity of the nuclear factor Bbf. Biochem J 311(Pt 3):769–773
Bevilacqua MA, Faniello MC, Quaresima B, Tiano MT, Giuliano P, Feliciello A, Avvedimento VE, Cimino F, Costanzo F (1997) A common mechanism underlying the E1A repression and the cAMP stimulation of the H ferritin transcription. J Biol Chem 272:20736–20741
Bevilacqua MA, Faniello MC, Russo T, Cimino F, Costanzo F (1998) P/CAF/p300 complex binds the promoter for the heavy subunit of ferritin and contributes to its tissue-specific expression. Biochem J 335(Pt 3):521–525
Bresgen N, Ohlenschlager I, Fiedler B, Wacht N, Zach S, Dunkelmann B, Arosio P, Kuffner E, Lottspeich F, Eckl PM (2007) Ferritin—a mediator of apoptosis? J Cell Physiol 212:157–164
Burbassi S, Sengupta R, Meucci O (2010) Alterations of CXCR4 function in mu-opioid receptor-deficient glia. Eur J Neurosci 32:1278–1288
Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL (2010) Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem 285:7805–7817
Cairo G, Castrusini E, Minotti G, Bernelli-Zazzera A (1996) Superoxide and hydrogen peroxide-dependent inhibition of iron regulatory protein activity: a protective stratagem against oxidative injury. FASEB J 10:1326–1335
Callewaere C, Banisadr G, Rostene W, Parsadaniantz SM (2007) Chemokines and chemokine receptors in the brain: implication in neuroendocrine regulation. J Mol Endocrinol 38:355–363
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924
Chalasani SH, Sabelko KA, Sunshine MJ, Littman DR, Raper JA (2003) A chemokine, SDF-1, reduces the effectiveness of multiple axonal repellents and is required for normal axon path finding. J Neurosci 23:1360–1371
Chalasani SH, Sabol A, Xu H, Gyda MA, Rasband K, Granato M, Chien CB, Raper JA (2007) Stromal cell-derived factor-1 antagonizes slit/robo signaling in vivo. J Neurosci 27:973–980
Charo IF, Ransohoff RM (2006) The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 354:610–621
Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen LY (2004) Heterodimerization and cross-desensitization between the mu-opioid receptor and the chemokine CCR5 receptor. Eur J Pharmacol 483:175–186
Christova T, Templeton DM (2007) Effect of hypoxia on the binding and subcellular distribution of iron regulatory proteins. Mol Cell Biochem 301:21–32
Cook A, Hippensteel R, Shimizu S, Nicolai J, Fatatis A, Meucci O (2010) Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J Biol Chem 285:10563–10571
Davies J, Everall IP, Weich S, Glass J, Sharer LR, Cho ES, Bell JE, Majteny C, Gray F, Scaravilli F, Lantos PL (1998) HIV-associated brain pathology: a comparative international study. Neuropathol Appl Neurobiol 24:118–124
Devi LA (2001) Heterodimerization of G-protein-coupled receptors: pharmacology, signaling and trafficking. Trends Pharmacol Sci 22:532–537
Dziembowska M, Tham TN, Lau P, Vitry S, Lazarini F, Dubois-Dalcq M (2005) A role for CXCR4 signaling in survival and migration of neural and oligodendrocyte precursors. Glia 50:258–269
Faniello MC, Bevilacqua MA, Condorelli G, de Crombrugghe B, Maity SN, Avvedimento VE, Cimino F, Costanzo F (1999) The B subunit of the CAAT-binding factor NFY binds the central segment of the Co-activator p300. J Biol Chem 274:7623–7626
Faniello MC, Chirico G, Quaresima B, Cuda G, Allevato G, Bevilacqua MA, Baudi F, Colantuoni V, Cimino F, Venuta S, Avvedimento VE, Costanzo F (2002) An alternative model of H ferritin promoter transactivation by c-Jun. Biochem J 363:53–58
Faniello MC, Di Sanzo M, Quaresima B, Baudi F, Di Caro V, Cuda G, Morrone G, Del Sal G, Spinelli G, Venuta S, Costanzo F (2008) p53-mediated downregulation of H ferritin promoter transcriptional efficiency via NF-Y. Int J Biochem Cell Biol 40:2110–2119
Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, Adler MW, Rogers TJ (2008) Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the kappa-opioid receptor. J Neuroimmunol 197:114–123
Guo B, Yu Y, Leibold EA (1994) Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J Biol Chem 269:24252–24260
Guo B, Phillips JD, Yu Y, Leibold EA (1995) Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem 270:21645–21651
Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF (2001) Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience 102:555–563
Guyon A, Rovere C, Cervantes A, Allaeys I, Nahon JL (2005) Stromal cell-derived factor-1alpha directly modulates voltage-dependent currents of the action potential in mammalian neuronal cells. J Neurochem 93:963–973
Haile DJ, Rouault TA, Harford JB, Kennedy MC, Blondin GA, Beinert H, Klausner RD (1992) Cellular regulation of the iron-responsive element binding protein: disassembly of the cubane iron–sulfur cluster results in high-affinity RNA binding. Proc Natl Acad Sci USA 89:11735–11739
Hanson ES, Leibold EA (1998) Regulation of iron regulatory protein 1 during hypoxia and hypoxia/reoxygenation. J Biol Chem 273:7588–7593
Hauser KF, El-Hage N, Buch S, Nath A, Tyor WR, Bruce-Keller AJ, Knapp PE (2006) Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol 1:98–105
Hintze KJ, Theil EC (2006) Cellular regulation and molecular interactions of the ferritins. Cell Mol Life Sci 63:591–600
Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ (2004) Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 101:18117–18122
Johansson T, Elfverson M, Zhou Q, Nyberg F (2010) Allosteric modulation of the NMDA receptor by neurosteroids in rat brain and the impact of long term morphine administration. Biochem Biophys Res Commun 401:504–508
Khan MZ, Brandimarti R, Musser BJ, Resue DM, Fatatis A, Meucci O (2003) The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J Neurovirol 9:300–314
Khan MZ, Brandimarti R, Patel JP, Huynh N, Wang J, Huang Z, Fatatis A, Meucci O (2004) Apoptotic and antiapoptotic effects of CXCR4: is it a matter of intrinsic efficacy? Implications for HIV neuropathogenesis. AIDS Res Hum Retroviruses 20:1063–1071
Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, Meucci O (2008) The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ 15:1663–1672
Ko SW, Wu LJ, Shum F, Quan J, Zhuo M (2008) Cingulate NMDA NR2B receptors contribute to morphine-induced analgesic tolerance. Mol Brain Res 1:2
Koch T, Seifert A, Wu DF, Rankovic M, Kraus J, Borner C, Brandenburg LO, Schroder H, Hollt V (2009) mu-opioid receptor-stimulated synthesis of reactive oxygen species is mediated via phospholipase D2. J Neurochem 110:1288–1296
Kuriyama-Matsumura K, Sato H, Suzuki M, Bannai S (2001) Effects of hyperoxia and iron on iron regulatory protein-1 activity and the ferritin synthesis in mouse peritoneal macrophages. Biochim Biophys Acta 1544:370–377
Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM (1995) Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem 270:15285–15293
Lam CF, Chang PJ, Huang YS, Sung YH, Huang CC, Lin MW, Liu YC, Tsai YC (2008) Prolonged use of high-dose morphine impairs angiogenesis and mobilization of endothelial progenitor cells in mice. Anesth Analg 107:686–692
LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R 3rd, Grinberg A, Love P, Tresser N, Rouault TA (2001) Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 27:209–214
Lavi E, Kolson DL, Ulrich AM, Fu L, Gonzalez-Scarano F (1998) Chemokine receptors in the human brain and their relationship to HIV infection. J Neurovirol 4:301–311
Lee JH, Jang H, Cho EJ, Youn HD (2009) Ferritin binds and activates p53 under oxidative stress. Biochem Biophys Res Commun 389:399–404
Lee ST, Chu K, Jung KH, Yoon HJ, Jeon D, Kang KM, Park KH, Bae EK, Kim M, Lee SK, Roh JK (2010) MicroRNAs induced during ischemic preconditioning. Stroke 41:1646–1651
Li L, Fang CJ, Ryan JC, Niemi EC, Lebron JA, Bjorkman PJ, Arase H, Torti FM, Torti SV, Nakamura MC, Seaman WE (2010) Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci USA 107:3505–3510
Li M, Ransohoff RM (2008) Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol 84:116–131
Li Q, Shirabe K, Thisse C, Thisse B, Okamoto H, Masai I, Kuwada JY (2005) Chemokine signaling guides axons within the retina in zebrafish. J Neurosci 25:1711–1717
Li R, Luo C, Mines M, Zhang J, Fan GH (2006) Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem 281:37616–37627
Lin KF, Chang RC, Suen KC, So KF, Hugon J (2004) Modulation of calcium/calmodulin kinase-II provides partial neuroprotection against beta-amyloid peptide toxicity. Eur J Neurosci 19:2047–2055
Lu M, Grove EA, Miller RJ (2002) Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci USA 99:7090–7095
Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA (1998) Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA 95:9448–9453
MacKenzie EL, Tsuji Y (2008) Elevated intracellular calcium increases ferritin H expression through an NFAT-independent post-transcriptional mechanism involving mRNA stabilization. Biochem J 411:107–113
Mikhael M, Kim SF, Schranzhofer M, Soe-Lin S, Sheftel AD, Mullner EW, Ponka P (2006) Iron regulatory protein-independent regulation of ferritin synthesis by nitrogen monoxide. FEBS J 273:3828–3836
Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM (1991) Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci USA 88:4946–4950
Nath A (2010) Human immunodeficiency virus-associated neurocognitive disorder: pathophysiology in relation to drug addiction. Ann NY Acad Sci 1187:122–128
Nicolai J, Burbassi S, Rubin J, Meucci O (2010) CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis 1(4):e33
Patel JP, Sengupta R, Bardi G, Khan MZ, Mullen-Przeworski A, Meucci O (2006) Modulation of neuronal CXCR4 by the micro-opioid agonist DAMGO. J Neurovirol 12:492–500
Patel JR, McCandless EE, Dorsey D, Klein RS (2010) CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci USA 107:11062–11067
Pello OM, Martinez-Munoz L, Parrillas V, Serrano A, Rodriguez-Frade JM, Toro MJ, Lucas P, Monterrubio M, Martinez AC, Mellado M (2008) Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur J Immunol 38:537–549
Pereska Z, Dejanova B, Bozinovska C, Petkovska L (2007) Prooxidative/antioxidative homeostasis in heroin addiction and detoxification. Bratisl Lek Listy 108:393–398
Peterson PK, Molitor TW, Chao CC (1998) The opioid–cytokine connection. J Neuroimmunol 83:63–69
Pitcher J, Shimizu S, Burbassi S, Meucci O (2010) Disruption of neuronal CXCR4 function by opioids: preliminary evidence of ferritin heavy chain as a potential etiological agent in neuroAIDS. J Neuroimmunol 224:66–71
Ragozzino D, Renzi M, Giovannelli A, Eusebi F (2002) Stimulation of chemokine CXC receptor 4 induces synaptic depression of evoked parallel fibers inputs onto Purkinje neurons in mouse cerebellum. J Neuroimmunol 127:30–36
Recalcati S, Invernizzi P, Arosio P, Cairo G (2008) New functions for an iron storage protein: the role of ferritin in immunity and autoimmunity. J Autoimmun 30:84–89
Robin AM, Zhang ZG, Wang L, Zhang RL, Katakowski M, Zhang L, Wang Y, Zhang C, Chopp M (2006) Stromal cell-derived factor 1alpha mediates neural progenitor cell motility after focal cerebral ischemia. J Cereb Blood Flow Metab 26:125–134
Rogers TJ, Peterson PK (2003) Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol 24:116–121
Rogers TJ, Steele AD, Howard OM, Oppenheim JJ (2000) Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann NY Acad Sci 917:19–28
Sammarco MC, Ditch S, Banerjee A, Grabczyk E (2008) Ferritin L and H subunits are differentially regulated on a post-transcriptional level. J Biol Chem 283:4578–4587
Sargeant TJ, Miller JH, Day DJ (2008) Opioidergic regulation of astroglial/neuronal proliferation: where are we now? J Neurochem 107:883–897
Schalinske KL, Eisenstein RS (1996) Phosphorylation and activation of both iron regulatory proteins 1 and 2 in HL-60 cells. J Biol Chem 271:7168–7176
Sengupta R, Burbassi S, Shimizu S, Cappello S, Vallee RB, Rubin JB, Meucci O (2009) Morphine increases brain levels of ferritin heavy chain leading to inhibition of CXCR4-mediated survival signaling in neurons. J Neurosci 29:2534–2544
Shpyleva SI, Tryndyak VP, Kovalchuk O, Starlard-Davenport A, Chekhun VF, Beland FA, Pogribny IP (2010) Role of ferritin alterations in human breast cancer cells. Breast Cancer Res Treat 126:63–71
Storr HL, Kind B, Parfitt DA, Chapple JP, Lorenz M, Koehler K, Huebner A, Clark AJ (2009) Deficiency of ferritin heavy-chain nuclear import in triple a syndrome implies nuclear oxidative damage as the primary disease mechanism. Mol Endocrinol 23:2086–2094
Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OM, Oppenheim JJ, Rogers TJ (2003) Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol 74:1074–1082
Theil EC (2007) Coordinating responses to iron and oxygen stress with DNA and mRNA promoters: the ferritin story. Biometals 20:513–521
Thomson AM, Rogers JT, Leedman PJ (1999) Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int J Biochem Cell Biol 31:1139–1152
Torti FM, Torti SV (2002) Regulation of ferritin genes and protein. Blood 99:3505–3516
Torti SV, Kwak EL, Miller SC, Miller LL, Ringold GM, Myambo KB, Young AP, Torti FM (1988) The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J Biol Chem 263:12638–12644
Toth I, Yuan L, Rogers JT, Boyce H, Bridges KR (1999) Hypoxia alters iron-regulatory protein-1 binding capacity and modulates cellular iron homeostasis in human hepatoma and erythroleukemia cells. J Biol Chem 274:4467–4473
Tran PB, Miller RJ (2003) Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci 4:444–455
Tsuji Y (2005) JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene 24:7567–7578
Tsuji Y, Moran E, Torti SV, Torti FM (1999) Transcriptional regulation of the mouse ferritin H gene. Involvement of p300/CBP adaptor proteins in FER-1 enhancer activity. J Biol Chem 274:7501–7507
Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM (2000) Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol 20:5818–5827
Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, Bruce-Keller AJ (2009) Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem 108:202–215
Vergote D, Butler GS, Ooms M, Cox JH, Silva C, Hollenberg MD, Jhamandas JH, Overall CM, Power C (2006) Proteolytic processing of SDF-1alpha reveals a change in receptor specificity mediating HIV-associated neurodegeneration. Proc Natl Acad Sci USA 103:19182–19187
Wallander ML, Zumbrennen KB, Rodansky ES, Romney SJ, Leibold EA (2008) Iron-independent phosphorylation of iron regulatory protein 2 regulates ferritin during the cell cycle. J Biol Chem 283:23589–23598
Wang W, Di X, Torti SV, Torti FM (2010a) Ferritin H induction by histone deacetylase inhibitors. Biochem Pharmacol 80:316–324
Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV (2010b) Serum ferritin: past, present and future. Biochim Biophys Acta 1800:760–769
Weber M, Modemann S, Schipper P, Trauer H, Franke H, Illes P, Geiger KD, Hengstler JG, Kleemann WJ (2006) Increased polysialic acid neural cell adhesion molecule expression in human hippocampus of heroin addicts. Neuroscience 138:1215–1223
Wei Y, Miller SC, Tsuji Y, Torti SV, Torti FM (1990) Interleukin 1 induces ferritin heavy chain in human muscle cells. Biochem Biophys Res Commun 169:289–296
Woeller CF, Fox JT, Perry C, Stover PJ (2007) A ferritin-responsive internal ribosome entry site regulates folate metabolism. J Biol Chem 282:29927–29935
Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM, Power C (2003) HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci 6:1064–1071
Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR (1998) Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393:595–599
Zumbrennen KB, Wallander ML, Romney SJ, Leibold EA (2009) Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Mol Cell Biol 29:2219–2229
Acknowledgements
The authors thank members of the Meucci Lab for helpful discussion and the NIH for generous support (R01-DA19808 and R01-DA15014 to OM). Anna (Cook) Abt is a fellow of the “Interdisciplinary and Translational Research Training in neuroAIDS” (T32-MH078795); thus, this investigation was supported in part by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award 5T32MH079785. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Conflicts of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Support
This work was funded by NIH grants R01-DA19808 and R01-DA15014 to Olimpia Meucci. Anna Abt is a fellow of the “Interdisciplinary and Translational Research Training in neuroAIDS” (T32-MH078795); thus, this investigation was also supported by the National Institutes of Health under the Ruth L. Kirschstein National Research Service Award 5T32MH079785.
Rights and permissions
About this article
Cite this article
Abt, A.C., Meucci, O. Regulation of Neuronal Ferritin Heavy Chain, A New Player in Opiate-Induced Chemokine Dysfunction. J Neuroimmune Pharmacol 6, 466–476 (2011). https://doi.org/10.1007/s11481-011-9278-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-011-9278-3