
Abstract
The development of novel immune-based therapeutics for neurodegenerative diseases is an area of intense focus. Neurodegenerative diseases represent a particular challenge since in many cases the onset of symptoms occurs after considerable degeneration has ensued. Based on human genetic and histopathological evidence from patients with neurodegenerative diseases, animal models that recapitulate specific pathologic features have been developed. Utilizing these animal models in combination with viral vector-based gene therapeutics, specific epochs of disease can be targeted. One common feature of several neurodegenerative diseases is misfolded proteins. The mechanism by which these altered protein conformers lead to neurodegeneration is not completely understood but much effort has been put forward to either degrade aberrant protein or prevent the formation of misfolded conformers. In this review, we will summarize work that employs viral vector gene therapeutics to modulate the brain’s response to misfolded proteins with a specific focus on neurodegeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases present a therapeutic challenge owing to the privileged environment afforded by the blood–brain barrier as well as delayed recognition of progressive diseases that begins insidiously. Most neurodegenerative diseases are diagnosed only after substantial neuronal or synaptic loss has occurred; thus precluding effective recovery and making symptomatic improvements transient. Neurodegenerative disorders have no cure and, for the majority of conditions, the causative agent(s) or etiologic factors are unknown. Therefore, most therapies are oriented toward symptomatic relief. A future in which disease progression can be managed mandates that neurodegenerative diseases be detected early and their mechanistic bases be understood; this includes the expansion of focused gene therapeutic approaches for restoration of function (Maguire-Zeiss and Federoff 2004; Maguire-Zeiss et al. 2007).
Gene therapy encompasses the delivery of genes to enhance function, to modulate disease progression, and/or to replace a defective gene. In the case of neurodegenerative diseases, enhancing neuronal function or “teaching old neurons new tricks” is exemplified by the use of nerve growth factor (NGF) for Alzheimer’s disease (AD) therapy. NGF is a member of the neurotrophin family and was the first nervous system growth factor identified (Levi-Montalcini and Hamburger 1951). Direct infusion of NGF into animals proved useful for the prevention of neuronal loss following lesion or traumatic injury, for promotion of neurite outgrowth, and for the reversal of age-related atrophy of basal forebrain cholinergic neurons (Hefti et al. 1984; Hefti 1986; Williams et al. 1986; Fischer et al. 1987; Kromer 1987; Sofroniew et al. 1990; Tuszynski 2007). Methods were then developed for ex vivo and in vivo gene therapy in an attempt to both target and restrict NGF expression to the appropriate brain regions. Clinical trials using ex vivo Moloney leukemia retrovirus encoding human nerve growth factor (MLV-NGF) transduced autologous fibroblasts that secrete NGF following stereotaxic neurosurgical implantation demonstrated safety while efficacy trials will require a larger number of subjects (Tuszynski and Gage 1990; Tuszynski 2007). Likewise, in vivo gene therapy using recombinant adeno-associated virus (rAAV) encoding NGF has been developed and a phase I clinical trial is underway (Bishop et al. 2008). In both cases, the expected outcome is a trophic response in cholinergic neurons with the attendant augmentation of cholinergic axon sprouting. Since these therapies rely on the presence of functioning neurons and do not address the disease mechanism, the effectiveness will likely wane as neurons succumb to the disease process. However, for slowly progressive age-related neurodegenerative diseases, gene therapy aimed at slowing disease elaboration can provide important symptom palliation and convert a fatal disease into a chronic disorder.
Curing disease is the fundamental goal of clinical medicine, and the field of gene therapy is focused in that direction. Replacement of a defective gene to cure disease is the ultimate objective of several gene therapeutic approaches. Perhaps the most well-known replacement gene therapy study was the targeting of severe combined immunodeficiency-X1 (SCID), an X-linked inherited disorder (Cavazzana-Calvo et al. 2000). Using a defective γc (IL2RG) Moloney retrovirus-derived vector combined with ex vivo infection of autologous CD34+ cells, the SCID phenotype was cured in nine out of the ten initially treated patients. However, enthusiasm for gene therapeutic approaches was dampened when several children developed acute lymphocytic leukemia presumably due to proviral integration within the LMO-2 gene locus which had previously been associated with human T cell acute lymphocytic leukemia (Rabbitts et al. 1999; Hacein-Bey-Abina et al. 2003). In spite of this adverse effect, the field is moving forward with cautious optimism.
In this review, we highlight the potential use of immune-directed gene therapeutic approaches to three neurodegenerative disorders with the common hallmark of misfolded toxic protein conformers: AD, prion disease, and Parkinson’s disease (PD). Targeting toxic protein conformations is an emerging field of neurotherapeutics and has broad applicability for other disorders as well. Specifically, we discuss the use of two viral-based systems, helper-free herpes simplex virus (HSV) amplicons and rAAV vectors, for the delivery of genes to engender a constructive immune response aimed at reducing toxic protein burden. Although this review does not cover issues regarding tropism and immunogenicity, both HSV amplicons and rAAV vectors hold promise for human gene therapy trials because of their low immunogenicity and tropism for specific cell types (Olschowka et al. 2003; Burger et al. 2004; Shevtsova et al. 2005; Cuchet et al. 2007; Taymans et al. 2007; Cearley et al. 2008; Mochizuki et al. 2008; Ulusoy et al. 2008).
Neuroimmune gene therapy for Alzheimer’s disease
AD is an age-related neurodegenerative disorder typified by progressive functional decline in executive functions leading to dementia and ultimately death (Yankner et al. 2008). Although symptoms vary considerably between patients, most often, affected individuals first present to the clinic with the most common symptom: difficulty remembering recently learned information. The memory impairment progresses until patients are unable to attend to activities of daily living and eventually die. In addition to symptoms associated with memory loss, some patients display neuropsychiatric manifestations including aggression and delusions. This broad-spectrum disorder is individualized with patients progressing at varying and unpredictable rates. Currently, there is no known etiology or cure for sporadic AD and, as the current population ages, it remains an enormous public health concern. Pathological evidence reveals that this disease is typified by the loss of cholinergic neurons and the presence of extracellular amyloid β (Aβ) plaques and tau-enriched neurofibrillary tangles (Hardy et al. 1998). Aβ deposits arise from amyloid precursor protein (APP) following a succession of protease-specific cleavages resulting in the small Aβ peptide (Hardy and Allsop 1991; Selkoe 1996; Hardy 1997; Hardy et al. 1998). Aβ following a nucleation event forms oligomers and finally the hallmark extracellular plaques and is thought to be responsible for the subsequent loss of cholinergic function.
Currently, most clinical therapeutic approaches for AD utilize cholinesterase inhibitors to manage symptoms by increasing cholinergic tone as well as other cognition enhancers (Bartus et al. 1982; Grutzendler and Morris 2001; Barten and Albright 2008). New potential areas for therapeutics are focused on decreasing the amount of toxic Aβ through inhibition of the pathogenic APP cleavage and, more recently, by vaccination approaches (Morgan et al. 2000; El-Amouri et al. 2008; Nalivaeva et al. 2008; Wolfe 2008). Immunotherapy is based on two general vaccination approaches, active and passive immunization (Brody and Holtzman 2008). Active vaccination requires exposure to the target antigen and production of antigen-specific antibodies following the activation of the cellular limb of the immune system. In contrast, for passive vaccination, the antibody or antibody-encoding gene is delivered directly, independent of cell-mediated immune activation. For neurodegenerative diseases, the goal of both vaccination approaches is an antibody-directed reduction in the amount of toxic misfolded protein and amelioration the disease.
The first report utilizing active immunization with aggregated Aβ in a mouse model of AD was met with great enthusiasm as this approach resolved the AD-like plaque pathology (Schenk et al. 1999; for review, see Brody and Holtzman 2008). Others soon demonstrated an attenuation of behavioral deficits as well as changes in plaque burden with behavioral improvements occurring even when aggregated Aβ was still present (Hsiao et al. 1996; Janus et al. 2000; Morgan et al. 2000; Chishti et al. 2001). These studies showed that behavioral improvement could be dissociated from complete elimination of plaque burden suggesting that vaccination might be successful if only a proportion of the Aβ plaque was removed. Alternatively, elimination of another pool of Aβ such as soluble (nonplaque-associated) Aβ might be responsible for the improved function in these treated animals. In either case, active vaccination with human aggregated Aβ (AN1792) and adjuvant soon moved into a phase I human clinical trial where safety, tolerability, and immunogenicity were confirmed (Bayer et al. 2005). Later, during the phase II clinical trial, adverse effects became evident with 6% of immunized patients developing aseptic meningoencephalitis and the trial was suspended. However, upon clinical examination of recruits, there was a trend toward decreased cognitive decline in the antibody responder group (19.7% patients) and a decrease in Aβ deposits in the brain’s of the few patients that had come to autopsy (Nicoll et al. 2003; Gilman et al. 2005; Bombois et al. 2007; Klunk et al. 2007; Petrushina et al. 2007; Brody and Holtzman 2008). These data gave hope that a vaccine approach for the treatment of AD was achievable. However, it was apparent that modulation of the immune response was needed since the AN1792 approach was not intended to modulate the type of immune response elicited.
The mechanism of removal of Aβ plaques following conventional peptide and adjuvant vaccination is not completely understood. On one hand, following immunization, a humoral response could ensue, efficiently clearing Aβ from the brain to the peripheral blood (DeMattos et al. 2001; Federoff and Bowers 2005). Conversely, anti-Aβ antibody fragment crystallizable (Fc) receptor-mediated activation of microglia could engender a phagocytotic response dissolving the Aβ plaques (Bard et al. 2000). It is possible that these two mechanisms act in collaboration to effect the immune response. Furthermore, Aβ is a self-peptide thus requiring coadministration of an adjuvant to elicit a strong immune response to break tolerance. This then begs the question: could Aβ active vaccination lead to an autoimmune condition? Furthermore, are varied responses to Aβ vaccination due to the underlying inflammatory state of the AD brain? The current vaccination approach would be considerably improved with the use of technologies that target and activate desired subsets of immune cell populations in an effort to increase degradation of Aβ while dampening deleterious CNS proinflammatory events (Federoff and Bowers 2005).
The causes of the adverse effects from the AN1792 vaccination trial are unclear. Presumably, when the fibrillar Aβ and coadministered adjuvant were taken up by antigen-presenting cells (APC) with the concomitant activation of the major histocompatibility complex II (MHC II) pathway and T cell engagement, the resultant adaptive immune response proceeded through the T cell helper 1 (TH1)-dependent pathway. The TH1 pathway led to the production of a subset of cytokines and chemokines (interferon-γ) that engendered B cell production of IgG2b-type anti-Aβ antibodies and perhaps activation of Aβ-specific cytotoxic T lymphocytes (CTL) resulting in a substantial and unwanted proinflammatory response. This activation of the cellular immune response could result in a dangerous perpetuating autoimmune response. The ideal Aβ-based vaccine would stimulate an immune response specifically against pathogenic Aβ devoid of cytotoxic T cell involvement resulting in dissolution of Aβ-containing aggregates without potentiation of brain inflammation. In essence, a vaccine that modulates the immune response to engender the humoral TH2 pathway leading to production of cytokines (i.e., interleukin-4) that do not promote inflammation and B cell-derived IgG1-type anti-Aβ antibodies decreasing the overall encephalitic potential (Federoff and Bowers 2005).
Modulating the immune system requires the delivery of genes or proteins that will be efficiently presented to effect the desired immune response. The HSV amplicon represents a versatile plasmid-based vector platform with a large insert payload (∼130 kb) and capable of efficient delivery of genes including immunomodulatory genes (Kutubuddin et al. 1999; Maguire-Zeiss et al. 2001; Tolba et al. 2001). In addition, HSV amplicons can transduce a number of cell types and, as such, they have proven to be multifunctional immunogens. Following HSV amplicon transduction of host stromal cells (i.e., fibroblasts), these peripheral cells provide a source of large amounts of antigen which, once released, are then cross-presented by professional DCs. In addition, HSV amplicons can directly transduce APC (DC) providing an efficient means of initiating an immune response (Santos et al. 2007). DCs migrate to the lymph nodes and efficiently present antigen to T cells. In addition, following migration to the lymph nodes, DCs can undergo apoptosis releasing antigen providing another source for cross priming. The overall result is efficient activation and expansion of T cells.
Employing HSV amplicon technology, Bowers and Federoff inoculated transgenic mice expressing mutated APP (Tg2576) with HSV amplicons encoding either Aβ1–42 (HSVAβ) or Aβ1–42 fused with the molecular adjuvant tetanus toxin Fragment C (HSVAβ/TtxFC) (Bowers et al. 2005). TtxFC was employed because it had been shown to enhance the immunogenicity and skew the immune response toward the humoral TH2 pathway (Lu et al. 1994). The Tg2576 mice vaccinated with HSVAβ/TtxFC generated IgG1 antibodies consistent with TH2 activation compared with HSVAβ-vaccinated mice which initially expressed IgM antibodies followed by IgA isotypes (Bowers et al. 2005). Overall, the HSVAβ/TtxFC-inoculated mice exhibited a humoral response to Aβ and reduced CNS Aβ deposits. However, the HSVAβ vaccine resulted in a surprising and marked increase in brain inflammation with upregulation of proinflammatory cytokines (TNFα, INFγ) that was uniquely toxic; death occurred 1–2 weeks after the second inoculation (Bowers et al. 2005). In a second study, this group further “shaped” the immune response by developing an HSV amplicon vector that expressed both Aβ and the pro-TH2 cytokine, interleukin-4 (IL-4; HSVIEAβCMVIL-4) (Frazer et al. 2008). In these studies, triple-transgenic AD mice (3xTg-AD) were employed since they develop both amyloid and neurofibrillary tangle pathology (Oddo et al. 2003). Animals inoculated with HSVIEAβCMVIL-4 demonstrated an increased TH2 response (more expression of IgG1 anti-Aβ antibodies), improved performance on a memory task, prevention of Aβ plaques, and decreased phosphorylated tau compared with nonvaccinated controls and animals vaccinated with HSV amplicon expressing Aβ alone (Frazer et al. 2008). The strength of this technology is evidenced by the codelivery of IL-4, which efficiently differentiated CD4+ T cells into the TH2 lineage. These data demonstrate the usefulness of HSV amplicons with their large DNA capacity to deliver multiple genes that modulate the immune response engendering a TH2 humoral response in the absence of apparent deleterious proinflammatory events. DNA delivery to modulate immune response is a therapeutic goal for many diseases portending the broad usefulness of HSV amplicon technology. For example, in multiple sclerosis (MS) clinical trials, DNA vaccination against myelin basic protein is being employed to tolerize in an antigen-specific manner and dampen the autoimmune process (Garren 2008; Garren et al. 2008). This therapy might be even more efficacious if the myelin basic protein DNA is codelivered with modulators of the TH1 response via HSV amplicon technology allowing for a more flexible modulation of the immune response.
Passive immunotherapy is also being investigated as a possible treatment for AD. First attempted with the direct infusion of monoclonal antibodies against Aβ into transgenic mouse models of AD, these studies demonstrated clear behavioral improvement that was again not directly linked to loss of aggregated Aβ (for review, see Brody and Holtzman 2008; Bard et al. 2000; DeMattos et al. 2001; Dodart et al. 2002; Kotilinek et al. 2002). It is possible that these antibodies recognize the unstable yet highly toxic oligomeric form of Aβ. An important safety concern that emerged from these rodent studies was the increased development of cerebral hemorrhages due to cerebral amyloid angiopathy (CAA) in antibody-treated APP transgenic mice (Pfeifer et al. 2002; Wilcock et al. 2004b; Racke et al. 2005). Another central issue is whether behavioral impairments in mouse models of AD accurately reflect AD patient behavioral deficits, advocating that therapies developed in rodent models require stepwise, careful studies in nonhuman primate models prior to clinical trial investigation. Extending similar therapies to other disease models will provide critical additional information regarding safety and efficacy. For instance, AD immunotherapies are being extended to other diseases with extracellular amyloid deposits. An example is age-related macular degeneration (AMD), a progressive retinal disease with similar AD pathological features including Aβ deposits where Ding et al. (2008) have demonstrated that systemic administration of anti-Aβ antibody resulted in decreased Aβ deposits in the retina and brain of an AMD mouse model.
Passive immunotherapy is not limited to monoclonal antibodies as demonstrated by Bacskai et al. (2002) following in vivo application of F(ab′)2 fragments of an anti-Aβ antibody which retains the antigen-binding site without the Fc receptor-mediated activation of microglia and macrophages resulting in a 45% clearance of Aβ deposits. However, Wilcock et al. (2004a, b), using F(ab′)2 fragments from a different antibody, failed to clear dense plaques but rather reported a reduction in diffuse plaque clearance. The use of the complete IgG demonstrated efficient removal of fibrillar Aβ with concomitant activation of microglia suggesting that, in this case, microglial activation may be critical for plaque clearance (Wilcock et al. 2004a). The success of a subset of F(ab′)2 fragments portends the usefulness of another modified antibody technology, single-chain antibodies which are discussed in the prion disease and PD sections of this review.
Passive vaccination as a gene therapy for the treatment of prionoses
Prionoses are a group of invariably fatal diseases commonly referred to as transmissible spongiform encephalopathies (TSE) and typified by the conversion of a cellular prion protein (PrPC) into a misfolded toxic protein (scrapie form; PrPSc). TSE exist as both familial and sporadic forms. The human variants include kuru, Creutzfeldt–Jakob disease (iatrogenic, new variant, familial, and sporadic CJD), Gerstmann–Straussler–Scheinker disease, fatal familial insomnia, and sporadic fatal insomnia while a number of other mammals are affected by the most well-known including scrapie (sheep), bovine spongiform encephalopathy (cattle), and chronic wasting disease (mule deer and elk; reviewed in Prusiner 2001). Each TSE has varied clinical and pathological features; however, the common thread is the conversion of PrPC to PrPSc that propagates the disease. There are no available treatments for TSEs.
The conversion of PrPC to PrPSc is incompletely understood but is believed to occur through direct interaction of the PrPSc with the PrPC. This is the first group of diseases identified that is propagated by protein–protein interactions and completely devoid of nucleic acids (Prusiner 2001). PrPC is enriched in both the lymphoreticular and central nervous system and is tethered to the extracellular surface of cells where it is available to interact with PrPSc and be converted to the pathological conformation. The extracellular location of PrPC portends the potential use of passive antibody therapy. Anti-PrPC antibody interaction with the cellular prion protein could prevent the conversion to PrPSc or alternatively enhance degradation of PrPC, in both cases depleting the pool of PrPC resulting in an overall decrease of the toxic protein. Enari et al. (2001) demonstrated that monoclonal antibodies against PrPC were capable of preventing PrPSc formation in susceptible cell lines demonstrating that loss of PrPC can disrupt disease propagation. Prionoses antibody therapy has been extended to single-chain antibodies (scFv) which are engineered antigen-binding molecules comprised of antigen-binding variable heavy (VH) and light (VL) chains linked by a small synthetic flexible peptide linker maintaining an active antigen-binding site (Malone and Sullivan 1996; Haidaris et al. 2001). scFvs are small polypeptides (∼25 kDa) for ready tissue penetration and produced from a single coding sequence so they can undergo facile molecular manipulation for expression from a variety of viral vectors. With this in mind, the monoclonal antibody employed by Enari et al. (2001) was converted to a scFv and found to be capable of binding PrPC and of efficient clearing of PrPSc in cultured cells (Donofrio et al. 2005).
A clinically relevant gene therapeutic passive immunization approach to prion disease requires long-term antibody expression if the mechanism of action is dependent upon direct binding of the antibody with cellular prion (PrPC). With this in mind, Wuertzer et al. (2008) utilized recombinant adeno-associated virus type 2 (rAAV2) delivery of anti-PrPC scFvs in a mouse model of TSE. rAAV was chosen because this vector is nonpathogenic to humans and demonstrates long-term expression with no discernable inflammatory side effects (Rabinowitz and Samulski 2000; Maguire-Zeiss and Federoff 2004). Three novel anti-PrPC scFvs were identified following screening of a phagemid library of human scFvs with PrPC (Malone and Sullivan 1996; Wuertzer et al. 2008). In addition, a scFv version of D18, a previously identified Fab antibody capable of reducing PrPSc burden, and a control scFv were utilized (Malone and Sullivan 1996; Peretz et al. 2001). All scFvs contained a murine immunoglobulin κ-secretory signal for efficient secretion following rAAV delivery. Mice were intracerebrally administered the rAAVscFv vectors (9 × 109 expression units, bilaterally), subsequently challenged with peripherally delivered infectious prions, and evaluated for therapeutic efficacy. rAAV-expressed anti-PrPC scFvs delayed the onset of prion pathogenesis as measured by clinical evaluation, rotarod performance, and decreased PrPSc burden (Wuertzer et al. 2008). The anti-PrPC scFvs with the highest affinity for the cognate antigen as measured by surface plasmon resonance was scFvD18 (bound PrPC 14 times more tightly than the other scFvs tested), which was also the most therapeutically effective. However, there was no direct correlation with binding affinity as one scFv was capable of binding PrPC but had no therapeutic effect suggesting that epitope-specific interactions are also important for efficacy. The therapeutic effects seen with a subset of anti-PrP scFvs implies that, once secreted, these scFvs were available to bind PrPC and prevent its misfolding into the toxic proteinase K-resistant PrPSc. While this treatment delayed the onset of disease, it was not curative, making clear the need for improvements. These might include enhancement of rAAV vector delivery to increase the number of transduced cells and, therefore, the total amount of secreted scFv, scFv maturation to augment antibody binding affinity, and viral titer optimization. However, these preliminary studies highlight the potential usefulness of the rAAV vector platform in combination with anti-PrPC scFvs for future prion immunotherapeutics.
Single-chain antibody-directed gene therapy for Parkinson’s disease
PD is an incurable age-related progressive neurodegenerative disorder with invariant loss of substantia nigra dopamine neurons (DANs). The loss of the neurotransmitter dopamine (DA) results in the common clinical motoric features of the disease including resting tremor, bradykinesia, rigidity, and postural instability. Hallmark pathological features of the disease include dystrophic projections to the striatum, intracytoplasmic protein inclusions called Lewy bodies, and activated microglia in addition to the paucity of DANs (McGeer et al. 1988; Spillantini et al. 1997, 1998; Ouchi et al. 2005; McGeer and McGeer 2008). The etiology of sporadic PD is not known but the role of the presynaptic protein α-synuclein (SYN) has been implicated by many as a contributor to pathogenesis (Maguire-Zeiss and Federoff 2003).
The normal function of SYN is unknown but because it is enriched in presynaptic terminals, binds lipid membranes, and is upregulated during song learning in zebra finches, it is hypothesized to be important for synaptic plasticity (George et al. 1995; Sidhu et al. 2004b; Bonini and Giasson 2005). The toxicity associated with SYN in human disease as well as in animal and cell models of PD points to protein misfolding as an important disease mediator. SYN lacks a secondary structure and readily misfolds into toxic oligomeric conformations consisting of β-sheets when exposed to changes in pH, molecular crowding, oxidative stress, and interactions with highly reactive molecules such as dopamine (Souza et al. 2000; Paxinou et al. 2001; Uversky et al., 2001a, b, c; Li et al. 2004; Rochet et al. 2004; Cappai et al. 2005; Maguire-Zeiss et al. 2005; Fink 2006; Maguire-Zeiss et al. 2006; Moussa et al. 2008). The specific mechanism by which SYN oligomers promote toxicity is not well-understood but is thought to involve a toxic gain of function perhaps by increasing cellular oxidative stress, engendering microglial activation, or forming annular pores in membranes disrupting cell function (Conway et al. 2000, 2001; Volles et al. 2001; Su et al. 2008).
Current PD therapies are aimed at augmenting DA production, increasing the half-life, or altering downstream neurotransmitter systems to mitigate the effects of the dopaminergic cell loss (Maguire-Zeiss et al. 2007). Similar to AD and TSE, we suggest that future PD therapy should include viral vector-based antiprotofibril therapies, specifically, modalities aimed at decreasing the amount of toxic oligomeric protein. In the case of PD, diminished SYN oligomers could be attained by the reduction of the amount of monomeric SYN that evolves to toxic oligomers, by the elimination/degradation of toxic SYN oligomers, or by an acceleration of the conversion from oligomers to higher-order yet purportedly less-toxic aggregates. We propose that passive immunotherapy is a viable approach for PD, but it is important to note that the toxic protein, SYN, is almost exclusively an intracellular protein as opposed to Aβ, which is extracellular, or PrPC, which is tethered to the outside surface of cell membranes. However, as the function of SYN is not completely understood and, in culture models of SYN overexpression this protein is released, the existence of extracellular SYN and its role should not be discounted (Lee et al. 2005; Su et al. 2008).
Gene-directed immunotherapy that targets cytoplasmic SYN can be accomplished utilizing intracellular scFv also called intrabodies, which are scFv produced for cytoplasmic expression. Intrabodies are powerful molecular tools as they can be directed to defined subcellular compartments and are amenable to viral vector delivery. These therapeutic agents have been applied to a variety of disease models including infectious diseases, cancers, and neurodegenerative diseases (for review, see Kontermann 2004). Several laboratories including our own have isolated scFv that bind to specific regions of SYN (Emadi et al. 2004; Zhou et al. 2004; Maguire-Zeiss et al. 2006; Emadi et al. 2007; Lynch et al. 2008). Emadi et al. (2004) were the first to identify human scFv against SYN that inhibited the rate of protein aggregation and the formation of SYN oligomers in a cell-free system. Further analysis of an anti-SYN intrabody (D10) in a cell culture model demonstrated intracellular binding of SYN, stabilization of detergent-soluble SYN with concomitant reduction in detergent-insoluble SYN as well as amelioration of SYN-induced cell morphological abnormalities (Zhou et al. 2004).
Using monomeric, aggregated, and dopamine-modified SYN, we interrogated a human scFv phage display library for sequence and conformation-specific anti-SYN binders (Maguire-Zeiss et al. 2006). We identified one anti-SYN scFv that specifically recognized a region within the nonamyloid component (NAC) of SYN, an area important for β-sheet oligomeric SYN formation (scFv3; amino acids 71–85) (Maguire-Zeiss et al. 2006). Another recently identified NAC-specific scFv when expressed as an intrabody reduced intracellular SYN aggregation and toxicity which bodes well for the use of this class of scFv as therapeutic agents (Lynch et al. 2008). We also identified two additional scFv that bind to the C terminus of SYN (scFv14 recognizes amino acids 106–120 and scFv15 recognizes amino acids 117–131) (Maguire-Zeiss et al. 2006). Upon Western blot analyses of monomeric and aggregated SYN, scFv15 recognized monomeric SYN as well as SYN species ∼≥200 kDa but did not interact with smaller oligomers. In contrast, scFv3 recognized SYN conformers that range in size from 16 to ≥250 kDa including intermediate oligomeric forms. Emadi et al. (2007) recently identified an oligomer-specific anti-SYN scFv that both inhibited SYN aggregation and toxicity following treatment of SH-SY5Y with extracellular aggregated SYN that was treated with the oligomer-specific scFv. What remains to be determined is the effectiveness of these intrabodies in animal models of PD.
The exact role that SYN plays in the death of DANs is not completely understood nor is the unique susceptibility of these neurons in PD. Genetic and environmental models of PD suggest that an interplay between SYN and mediators of oxidative stress play an important part in the development of PD (Maguire-Zeiss and Federoff 2003; Perez and Hastings 2004; Sidhu et al. 2004a, b; Maguire-Zeiss et al. 2005). Since SYN is enriched in presynaptic DA terminals and DA is a highly reactive molecule capable of forming a quinone that readily modifies SYN engendering a misfolded state, it follows that methods to prevent DA-induced SYN misfolding could be novel therapeutic agents (Conway et al. 2001; Maguire-Zeiss et al. 2006). With that in mind, we sought to identify scFv that would recognize DA-modified SYN. Following panning of the human scFv phage display library with DA-modified SYN, we identified one scFv that recognizes DA-modified proteins exclusively (scFv6; Maguire-Zeiss et al. 2006). This DA-specific binder does not recognize monomeric, aggregated SYN, linear peptide SYN sequences, or DA alone but does bind to DA-modified proteins. This data supports the concept that this scFv is conformation-specific rather than sequence-specific.
These scFv represent a renewable and potentially powerful set of reagents that could be used as both diagnostic and therapeutic agents. For example, utilizing conformation-specific scFv, cerebrospinal fluid (CSF) and blood from PD patients could be interrogated for specific SYN conformers. In support of this application, using commercially available monoclonal antibodies in an enzyme-linked immunosorbent assay method SYN has been identified in both human plasma and CSF (El-Agnaf et al. 2006; Tokuda et al. 2006). However, the anti-SYN scFv and anti-DA-modified SYN scFv discussed in this review represent a relatively inexpensive and renewable source of antibody with the potential to more easily discriminate different conformers of SYN. As mentioned in our TSE discussion, these scFv are easily manipulated for both intrabody and secreted production in mammalian cells. The PD field currently awaits the use of these novel scFv in animal models of PD to determine their efficacy as therapeutic agents. We have molecularly modified our anti-SYN scFv for intracellular expression from both HSV amplicon and rAAV vectors and are currently testing them in an animal model of PD.
Conclusion
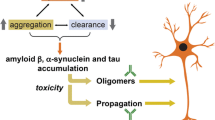
In this review, we discussed the use of active and passive immunization in animal models of neurodegenerative diseases highlighting work from our laboratory. The common thread for all of the neurodegenerative diseases presented is the presence of a toxic misfolded protein that seemingly drives the disease process. The misfolding pathway for each protein has not been completely elucidated but, in somewhat pedestrian terms, these proteins move from a monomeric or native conformation to an oligomeric protofibrillar toxic state finally to a large aggregate that is no longer toxic and in fact may be destined for degradation by cellular machinery (Fig. 1). We have described several immunotherapeutic methods to interfere with this pathway. For example, active immunization of an AD mouse with an HSV amplicon encoding the Aβ antigen and pro-TH2 cytokines dissolved the extracellular Aβ plaques and improved behavioral deficits in the absence of proinflammatory events (Fig. 1a). While gene-directed passive immunization of a prion mouse with rAAV encoding secreted anti-PrPC scFv decreased the conversion to the misfolded pathogenic scrapie form of prion protein and improved clinical outcome measurements (Fig. 1b). It remains to be determined whether delivering higher titers of rAAV will enhance the improved clinical outcome. Finally, several scFv directed at different forms of SYN have been identified and are capable of inhibiting aggregation. However, SYN is an interesting case because we can also imagine that scFv binding could result in accelerated aggregation of this protein since it is known to easily undergo conformational changes. Acceleration of SYN aggregate formation may actually prove to be protective since higher-order aggregates are posited to be nontoxic (Fig. 1c; Volles et al. 2001). Therefore, in our outlined pathway of protein misfolding, a reduction in the toxic protofibril could be accomplished by decreasing the input (monomeric protein), increasing protofibril degradation perhaps by the binding of intrabodies that direct the protein to the proteosome, or accelerating the production of larger aggregates. The area of immunotherapy for neurodegenerative diseases is just emerging, but with continued improvements in viral vector-regulated gene expression, enhanced delivery techniques, and the advent of peripheral biomarkers for early detection of these devastating diseases, the future promises to be very exciting.

Immunotherapy for neurodegenerative diseases. AD, TSE, and PD share a common hallmark pathobiologic feature of misfolded toxic proteins. In this review, we discuss the use of both active and passive vaccination approaches to decrease the amount of the toxic moieties. a Active vaccination utilizing HSV amplicon delivery of Aβ and IL-4 shaped the immune response toward TH2 in a mouse model of AD decreasing the Aβ plaque levels (Frazer et al. 2008). b Passive vaccination of scrapie-challenged mice with rAAVscFvPrPC decreased the PrPSc burden and improved clinical and behavioral outcome measurements (Wuertzer et al. 2008). c Anti-SYN scFv have been identified with preferential binding to monomeric and/or protofibrillar SYN conformers (Emadi et al. 2004, 2007; Zhou et al. 2004; Maguire-Zeiss et al. 2006; Lynch et al. 2008). Subsets of these scFv have demonstrable efficacy in preventing SYN aggregation (Emadi et al. 2004; Zhou et al. 2004; Emadi et al. 2007; Lynch et al. 2008). In this schematic, we also consider scFv that may accelerate the formation of toxic oligomers to larger nontoxic aggregates
References
Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT (2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci 22:7873–7878
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6:916–919. doi:10.1038/78682
Barten DM, Albright CF (2008) Therapeutic strategies for Alzheimer’s disease. Mol Neurobiol 37(2–3):171–186
Bartus RT, Dean RLd, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217:408–414. doi:10.1126/science.7046051
Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S (2005) Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64:94–101. doi:10.1159/000087764
Bishop KM, Hofer EK, Mehta A, Ramirez A, Sun L, Tuszynski M, Bartus RT (2008) Therapeutic potential of CERE-110 (AAV2-NGF): targeted, stable, and sustained NGF delivery and trophic activity on rodent basal forebrain cholinergic neurons. Exp Neurol 211:574–584. doi:10.1016/j.expneurol.2008.03.004
Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, Lavielle R, Delacourte A, Pasquier F (2007) Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol 64:583–587. doi:10.1001/archneur.64.4.583
Bonini NM, Giasson BI (2005) Snaring the function of alpha-synuclein. Cell 123:359–361. doi:10.1016/j.cell.2005.10.017
Bowers WJ, Mastrangelo MA, Stanley HA, Casey AE, Milo LJ Jr, Federoff HJ (2005) HSV amplicon-mediated Abeta vaccination in Tg2576 mice: differential antigen-specific immune responses. Neurobiol Aging 26:393–407. doi:10.1016/j.neurobiolaging.2004.04.006
Brody DL, Holtzman DM (2008) Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci 31:175–193. doi:10.1146/annurev.neuro.31.060407.125529
Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N (2004) Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther 10:302–317. doi:10.1016/j.ymthe.2004.05.024
Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF (2005) Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J 19:1377–1379
Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, Bousso P, Deist FL, Fischer A (2000) Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288:669–672. doi:10.1126/science.288.5466.669
Cearley CN, Vandenberghe LH, Parente MK, Carnish ER, Wilson JM, Wolfe JH (2008) Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol Ther 16(10):1710–1718
Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570. doi:10.1074/jbc.M100710200
Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A 97:571–576. doi:10.1073/pnas.97.2.571
Conway KA, Rochet JC, Bieganski RM, Lansbury PT Jr (2001) Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein Adduct. Science 294:1346–1349. doi:10.1126/science.1063522
Cuchet D, Potel C, Thomas J, Epstein AL (2007) HSV-1 amplicon vectors: a promising and versatile tool for gene delivery. Expert Opin Biol Ther 7:975–995. doi:10.1517/14712598.7.7.975
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti-Abeta antibody alters CNS and plasma Abeta clearance and decreases brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 3:3
Ding JD, Lin J, Mace BE, Herrmann R, Sullivan P, Bowes Rickman C (2008) Targeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an age-related macular degeneration mouse model. Vision Res 48:339–345. doi:10.1016/j.visres.2007.07.025
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 5:452–457
Donofrio G, Heppner FL, Polymenidou M, Musahl C, Aguzzi A (2005) Paracrine inhibition of prion propagation by anti-PrP single-chain Fv miniantibodies. J Virol 79:8330–8338. doi:10.1128/JVI.79.13.8330-8338.2005
El-Agnaf OM, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA, Schlossmacher MG, Allsop D (2006) Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 20:419–425. doi:10.1096/fj.03-1449com
El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS (2008) Neprilysin: an enzyme candidate to slow the progression of Alzheimer’s disease. Am J Pathol 172:1342–1354. doi:10.2353/ajpath.2008.070620
Emadi S, Liu R, Yuan B, Schulz P, McAllister C, Lyubchenko Y, Messer A, Sierks MR (2004) Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry 43:2871–2878. doi:10.1021/bi036281f
Emadi S, Barkhordarian H, Wang MS, Schulz P, Sierks MR (2007) Isolation of a human single chain antibody fragment against oligomeric alpha-synuclein that inhibits aggregation and prevents alpha-synuclein-induced toxicity. J Mol Biol 368:1132–1144. doi:10.1016/j.jmb.2007.02.089
Enari M, Flechsig E, Weissmann C (2001) Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A 98:9295–9299. doi:10.1073/pnas.151242598
Federoff HJ, Bowers WJ (2005) Immune shaping and the development of Alzheimer’s disease vaccines. Sci Aging Knowledge Environ 2005(46):pe35. doi:10.1126/sageke.2005.46.pe35
Fink AL (2006) The aggregation and fibrillation of alpha-synuclein. Acc Chem Res 39:628–634. doi:10.1021/ar050073t
Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH (1987) Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 329:65–68. doi:10.1038/329065a0
Frazer ME, Hughes JE, Mastrangelo MA, Tibbens JL, Federoff HJ, Bowers WJ (2008) Reduced pathology and improved behavioral performance in Alzheimer’s disease mice vaccinated with HSV amplicons expressing amyloid-beta and interleukin-4. Mol Ther 16:845–853. doi:10.1038/mt.2008.39
Garren H (2008) A DNA vaccine for multiple sclerosis. Expert Opin Biol Ther 8:1539–1550. doi:10.1517/14712598.8.10.1539
Garren H, Robinson WH, Krasulova E, Havrdova E, Nadj C, Selmaj K, Losy J, Nadj I, Radue EW, Kidd BA, Gianettoni J, Tersini K, Utz PJ, Valone F, Steinman L (2008) Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol 63:611–620. doi:10.1002/ana.21370
George JM, Jin H, Woods WS, Clayton DF (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15:361–372. doi:10.1016/0896-6273(95)90040-3
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64:1553–1562. doi:10.1212/01.WNL.0000159740.16984.3C
Grutzendler J, Morris JC (2001) Cholinesterase inhibitors for Alzheimer’s disease. Drugs 61:41–52. doi:10.2165/00003495-200161010-00005
Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A (2003) A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348:255–256. doi:10.1056/NEJM200301163480314
Haidaris CG, Malone J, Sherrill LA, Bliss JM, Gaspari AA, Insel RA, Sullivan MA (2001) Recombinant human antibody single chain variable fragments reactive with Candida albicans surface antigens. J Immunol Methods 257:185–202. doi:10.1016/S0022-1759(01)00463-X
Hardy J (1997) Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 20:154–159. doi:10.1016/S0166-2236(96)01030-2
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388. doi:10.1016/0165-6147(91)90609-V
Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M (1998) Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci 1:355–358. doi:10.1038/1565
Hefti F (1986) Nerve growth factor (NGF) promotes survival of septal cholinergic neurons after fimbrial transection. J Neurosci 6:2155–2162
Hefti F, Dravid A, Hartikka J (1984) Chronic intraventricular injections of nerve growth factor elevate hippocampal choline acetyltransferase activity in adult rats with partial septo-hippocampal lesions. Brain Res 293:305–311. doi:10.1016/0006-8993(84)91237-X
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi:10.1126/science.274.5284.99
Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D (2000) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408:979–982. doi:10.1038/35050110
Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST (2007) Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 27:6174–6184. doi:10.1523/JNEUROSCI.0730-07.2007
Kontermann RE (2004) Intrabodies as therapeutic agents. Methods 34:163–170. doi:10.1016/j.ymeth.2004.04.002
Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH (2002) Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci 22:6331–6335
Kromer LF (1987) Nerve growth factor treatment after brain injury prevents neuronal death. Science 235:214–216. doi:10.1126/science.3798108
Kutubuddin M, Federoff HJ, Challita-Eid PM, Halterman M, Day B, Atkinson M, Planelles V, Rosenblatt JD (1999) Eradication of pre-established lymphoma using herpes simplex virus amplicon vectors. Blood 93:643–654
Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016–6024. doi:10.1523/JNEUROSCI.0692-05.2005
Levi-Montalcini R, Hamburger V (1951) Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool 116:321–362. doi:10.1002/jez.1401160206
Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL (2004) Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson’s and Alzheimer’s disease. FASEB J 18:962–964. doi:10.1096/fj.04-2273com
Lu CH, Lee CJ, Kind P (1994) Immune responses of young mice to pneumococcal type 9V polysaccharide-tetanus toxoid conjugate. Infect Immun 62:2754–2760
Lynch SM, Zhou C, Messer A (2008) An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J Mol Biol 377:136–147. doi:10.1016/j.jmb.2007.11.096
Maguire-Zeiss KA, Federoff HJ (2003) Convergent pathobiologic model of Parkinson’s disease. Ann N Y Acad Sci 991:152–166
Maguire-Zeiss KA, Federoff HJ (2004) Safety of viral vectors for neurological gene therapies. Curr Opin Mol Ther 6:473–481
Maguire-Zeiss KA, Bowers WJ, Federoff HJ (2001) HSV vector-mediated gene delivery to the central nervous system. Curr Opin Mol Ther 3:482–490
Maguire-Zeiss KA, Short DW, Federoff HJ (2005) Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson’s disease? Brain Res Mol Brain Res 134:18–23. doi:10.1016/j.molbrainres.2004.09.014
Maguire-Zeiss KA, Wang CI, Yehling E, Sullivan MA, Short DW, Su X, Gouzer G, Henricksen LA, Wuertzer CA, Federoff HJ (2006) Identification of human alpha-synuclein specific single chain antibodies. Biochem Biophys Res Commun 349:1198–1205. doi:10.1016/j.bbrc.2006.08.127
Maguire-Zeiss KA, Mhyre TR, Federoff HJ (2007) Gazing into the future: Parkinson’s disease gene therapeutics to modify natural history. Exp Neurol 209(1):101–113
Malone J, Sullivan MA (1996) Analysis of antibody selection by phage display utilizing anti-phenobarbital antibodies. J Mol Recognit 9:738–745. doi:10.1002/(SICI)1099-1352(199634/12)9:5/6<738::AID-JMR333>3.0.CO;2-V
McGeer PL, McGeer EG (2008) Glial reactions in Parkinson’s disease. Mov Disord 23:474–483. doi:10.1002/mds.21751
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
Mochizuki H, Yasuda T, Mouradian MM (2008) Advances in gene therapy for movement disorders. Neurotherapeutics 5:260–269. doi:10.1016/j.nurt.2008.01.005
Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408:982–985. doi:10.1038/35050116
Moussa CE, Mahmoodian F, Tomita Y, Sidhu A (2008) Dopamine differentially induces aggregation of A53T mutant and wild type alpha-synuclein: insights into the protein chemistry of Parkinson’s disease. Biochem Biophys Res Commun 365:833–839. doi:10.1016/j.bbrc.2007.11.075
Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ (2008) Amyloid-degrading enzymes as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 5:212–224. doi:10.2174/156720508783954785
Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 9:448–452. doi:10.1038/nm840
Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging 24:1063–1070. doi:10.1016/j.neurobiolaging.2003.08.012
Olschowka JA, Bowers WJ, Hurley SD, Mastrangelo MA, Federoff HJ (2003) Helper-free HSV-1 amplicons elicit a markedly less robust innate immune response in the CNS. Mol Ther 7:218–227. doi:10.1016/S1525-0016(02)00036-9
Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T (2005) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 57:168–175. doi:10.1002/ana.20338
Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H (2001) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 21:8053–8061
Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB (2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412:739–743. doi:10.1038/35089090
Perez RG, Hastings TG (2004) Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J Neurochem 89:1318–1324. doi:10.1111/j.1471-4159.2004.02423.x
Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H, Patel A, Head E, Cribbs DH, Agadjanyan MG (2007) Alzheimer’s disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Abeta species in amyloid precursor protein transgenic mice. J Neurosci 27:12721–12731. doi:10.1523/JNEUROSCI.3201-07.2007
Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M (2002) Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 298:1379. doi:10.1126/science.1078259
Prusiner SB (2001) Shattuck lecture-neurodegenerative diseases and prions. N Engl J Med 344:1516–1526. doi:10.1056/NEJM200105173442006
Rabbitts TH, Bucher K, Chung G, Grutz G, Warren A, Yamada Y (1999) The effect of chromosomal translocations in acute leukemias: the LMO2 paradigm in transcription and development. Cancer Res 59:1794s–1798s
Rabinowitz JE, Samulski RJ (2000) Building a better vector: the manipulation of AAV virions. Virology 278:301–308. doi:10.1006/viro.2000.0707
Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB (2005) Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci 25:629–636. doi:10.1523/JNEUROSCI.4337-04.2005
Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT (2004) Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci 23:23–34. doi:10.1385/JMN:23:1-2:023
Santos K, Simon DA, Conway E, Bowers WJ, Mitra S, Foster TH, Lugade A, Lord EM, Federoff HJ, Dewhurst S, Frelinger JG (2007) Spatial and temporal expression of herpes simplex virus type 1 amplicon-encoded genes: implications for their use as immunization vectors. Hum Gene Ther 18:93–105. doi:10.1089/hum.2006.082
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177. doi:10.1038/22124
Selkoe DJ (1996) Amyloid beta-protein and the genetics of Alzheimer’s disease. J Biol Chem 271:18295–18298
Shevtsova Z, Malik JM, Michel U, Bahr M, Kugler S (2005) Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol 90:53–59. doi:10.1113/expphysiol.2004.028159
Sidhu A, Wersinger C, Vernier P (2004a) Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J 18:637–647. doi:10.1096/fj.03-1112rev
Sidhu A, Wersinger C, Vernier P (2004b) alpha-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett 565:1–5. doi:10.1016/j.febslet.2004.03.063
Sofroniew MV, Galletly NP, Isacson O, Svendsen CN (1990) Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science 247:338–342. doi:10.1126/science.1688664
Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H (2000) Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem 275:18344–18349. doi:10.1074/jbc.M000206200
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. doi:10.1038/42166
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. doi:10.1073/pnas.95.11.6469
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29:1690–1701
Taymans JM, Vandenberghe LH, Haute CV, Thiry I, Deroose CM, Mortelmans L, Wilson JM, Debyser Z, Baekelandt V (2007) Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther 18:195–206. doi:10.1089/hum.2006.178
Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, Locascio JJ, Schlossmacher MG, El-Agnaf OM (2006) Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun 349:162–166. doi:10.1016/j.bbrc.2006.08.024
Tolba KA, Bowers WJ, Hilchey SP, Halterman MW, Howard DF, Giuliano RE, Federoff HJ, Rosenblatt JD (2001) Development of herpes simplex virus-1 amplicon-based immunotherapy for chronic lymphocytic leukemia. Blood 98:287–295. doi:10.1182/blood.V98.2.287
Tuszynski MH (2007) Nerve growth factor gene therapy in Alzheimer disease. Alzheimer Dis Assoc Disord 21:179–189. doi:10.1097/WAD.0b013e318068d6d2
Tuszynski MH, Gage FH (1990) Potential use of neurotrophic agents in the treatment of neurodegenerative disorders. Acta Neurobiol Exp (Wars) 50:311–322
Ulusoy A, Bjorklund T, Hermening S, Kirik D (2008) In vivo gene delivery for development of mammalian models for Parkinson’s disease. Exp Neurol 209:89–100. doi:10.1016/j.expneurol.2007.09.011
Uversky VN, Li J, Fink AL (2001a) Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular link between Parkinson’s disease and heavy metal exposure. J Biol Chem 276:44284–44296. doi:10.1074/jbc.M105343200
Uversky VN, Li J, Fink AL (2001b) Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 276:10737–10744. doi:10.1074/jbc.M010907200
Uversky VN, Lee HJ, Li J, Fink AL, Lee SJ (2001c) Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem 276:43495–43498. doi:10.1074/jbc.C100551200
Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT Jr (2001) Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40:7812–7819. doi:10.1021/bi0102398
Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D (2004a) Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis 15:11–20. doi:10.1016/j.nbd.2003.09.015
Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D (2004b) Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation 1:24. doi:10.1186/1742-2094-1-24
Williams LR, Varon S, Peterson GM, Wictorin K, Fischer W, Bjorklund A, Gage FH (1986) Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc Natl Acad Sci U S A 83:9231–9235. doi:10.1073/pnas.83.23.9231
Wolfe MS (2008) Gamma-secretase inhibition and modulation for Alzheimer’s disease. Curr Alzheimer Res 5:158–164. doi:10.2174/156720508783954767
Wuertzer CA, Sullivan MA, Qiu X, Federoff HJ (2008) CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol Ther 16:481–486. doi:10.1038/sj.mt.6300387
Yankner BA, Lu T, Loerch P (2008) The aging brain. Annu Rev Pathol 3:41–66. doi:10.1146/annurev.pathmechdis.2.010506.092044
Zhou C, Emadi S, Sierks MR, Messer A (2004) A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed alpha-synuclein. Mol Ther 10:1023–1031. doi:10.1016/j.ymthe.2004.08.019
Conflict of Interest Disclosure
These authors have filed a U.S. Provisional Patent Application (“Alpha-synuclein antibodies and methods related there to”) based upon some of the work presented in this review. There are no other actual or potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors who are guarantors of the work: Kathleen A. Maguire-Zeiss and Howard J. Federoff. Source of support: NIEHS R01ES014470 (KMZ); DAMD17-03-1-0009 (HJF).
Rights and permissions
About this article
Cite this article
Maguire-Zeiss, K.A., Federoff, H.J. Immune-Directed Gene Therapeutic Development for Alzheimer’s, Prion, and Parkinson’s Diseases. J Neuroimmune Pharmacol 4, 298–308 (2009). https://doi.org/10.1007/s11481-008-9133-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-008-9133-3