
Abstract
Fungal co-culture is a strategy to induce the production of secondary metabolites by activating cryptic genes. We discovered the production of a new compound, talarodone A (1), along with five known compounds 2–6 in co-culture of Talaromyces pinophilus and Paraphaeosphaeria sp. isolated from soil collected in Miyazaki Prefecture, Japan. Among them, the productions of penicidones C (2) and D (3) were enhanced 27- and sixfold, respectively, by the co-culture. The structure of 3 should be represented as a γ-pyridol form with the reported chemical shifts, but not as a γ-pyridone form, based on DFT calculation.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many medical agents have been developed from natural products as a result of their novel skeletons and various bioactivities appropriate to drugs. Although more than half of the drugs approved by the FDA were developed from natural products [1], the number of antibiotics developed from microorganisms has decreased recently. Biosynthetic gene clusters of the majority of microorganisms (> 95%) remain cryptic under standard laboratory culture conditions, as revealed by recent metagenomic studies [2,3,4]. Efforts such as (1) alteration of growth conditions, (2) addition of chemical elicitors, (3) modification of targeted genes, (4) expression of genes in heterologous hosts, and (5) co-culturing have been employed to activate these cryptic genes to produce microbial metabolites [5, 6]. Microorganisms in co-cultures are considered to physiologically and chemically stimulate each other, leading to activation of the cryptic genes and production of metabolites that are not produced in mono-cultures [7, 8].
During a search for new drug candidates from cryptic fungal metabolites, we reported the isolation of a new indolinone derivative, irpexine, from the co-culture of Irpex lacteus (Fr.) Fr. (13S052) and Phaeosphaeria oryzae I. Miyake (13S070) [9]. In this study, we prepared 132 mutually co-cultured extracts using 12 fungi collected in Miyazaki Prefecture, Japan. Analysis by LC–MS indicated the presence of a new peak in the co-culture extract of Talaromyces pinophilus (17F4103) and Paraphaeosphaeria sp. (17F4110). We here report the isolation and structure determination of a new compound, talarodone A (1) (Fig. 1), by spectroscopic data and DFT calculation. In addition, among the five known compounds 2–6 (Fig. 1) isolated, the productions of penicidones C [10] (2) and D [11] (3) were enhanced by co-culture. During identification, we found that the structure of 3 should be represented as a γ-pyridol form (3a) with the reported chemical shifts, but not as a γ-pyridone form (3b), based on the DFT calculation.

Structures of compounds 1–6
Results and discussion
Separation of fungal metabolites 1–6
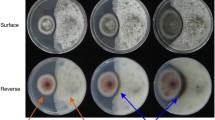
Two fungi, T. pinophilus (17F4103) and Paraphaeosphaeria sp. (17F4110), were inoculated on malt agar medium and cultured (Fig. 2a). LC–MS analysis of the extracts of the co-culture and mono-cultures revealed the presence of a new ion peak at m/z 406 ([M+H]+) in the co-culture that was absent in the mono-cultures (Fig. 2b). This result indicated that the co-culture activated the production of a new compound, 1.

a Co-culture of T. pinophilus (17F4103) and Paraphaeosphaeria sp. (17F4110). b LC–MS profiles of the extracts of the co-culture and mono-cultures for 1 extracted with m/z 406 [M+H]+
The MeOH extract of the co-culture was partitioned between EtOAc and H2O, and the aqueous layer was further partitioned between n-BuOH and H2O. The n-BuOH fraction containing the compound with m/z 406 was purified by ODS column chromatography and HPLC to afford 1. LC–MS analysis of the EtOAc fraction indicated the presence of congeners of 1. This was purified to afford penicidones C (2) and D (3), 3-O-methylfunicone [12] (4), ( ±)-penifupyrone [13] (5), and pinophilin B [14] (6).
Structure elucidation of new compound 1
The molecular formula of 1 was determined as C20H23NO8 (m/z 406.1528 [M+H]+). The 1H and 13C NMR data of 1 (Table 1) were similar to those of penicidon D (3), except for the presence of a 2-hydroxypropyl group (C1″–C3″) in 1 instead of a 1-propenyl group in 3, which was confirmed by COSY and HMBC correlations (Fig. 3). DFT calculation was applied for the γ-pyridone and γ-pyridol forms of 1 (Table 2). The standard deviations (SDs) of the differences between the observed and calculated shifts for the γ-pyridone and γ-pyridol forms were 2.5 and 7.0, respectively, which indicated 1 to be a γ-pyridone form. The differences of C-8 and C1′–C6′ in the γ-pyridol form were particularly large. Because the optical rotation of 1 was almost zero, − 1.8 (c = 0.33), 1 may be a racemate.

COSY and HMBC correlations of 1
Revision of structure description of penicidone D (3)
The structure of 3 was previously reported as a γ-pyridone form (3b), like 2, without any detailed analyses [11]. We found that the 13C signals at C-3′ (δ 153.7) and C-5′ (δ 147.9) (Table 3; our data) of 3 were shifted downfield, which is characteristic of the γ-pyridol form. We confirmed this by DFT calculation (Table 3). SDs of the differences between the observed and calculated shifts for the γ-pyridone and γ-pyridol forms clearly showed that 3 should be represented as a γ-pyridol form; notably, the differences of C-7, C-8, C2′–C6′, and C-2″ in the γ-pyridone form were large.
Conclusions
Co-culture of T. pinophilus (17F4103) and Paraphaeosphaeria sp. (17F4110) afforded a new metabolite, 1, along with five known compounds, 2–6. Among these compounds, the productions of 2 and 3 were enhanced 27- and sixfold, respectively, by the co-culture (Fig. 4), while those of 4–6 were not changed. It is noteworthy that the γ-pyridone/ γ-pyridol forms in 1–3 in an aprotic solvent are converted by the substituted groups. Among them, only 3 has a γ-pyridol form. Compared to 1 and 3, the substituents at C-3′ are different, i.e., a 2-hydroxypropyl group in 1 and a 1-propenyl group in 3, and thus 3 corresponds to a 2′-methoxy derivative of 2. The compounds isolated in this study did not show cytotoxicity or antimicrobial activity.

LC–MS profiles of the extracts of the co-culture and mono-cultures for 2 and 3 extracted with m/z 358 (a) and 388 (b), respectively
Experimental
General experimental procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter in 50% MeOH–H2O. UV spectra were measured on a JASCO V-550 spectrophotometer in 50% MeOH-H2O. IR spectra were recorded on a Perkin Elmer Frontier FT-IR spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker Avance III HD 500 NMR spectrometer in DMSO-d6 or acetone-d6. Chemical shifts were referenced to the residual solvent peaks (δH 2.49 and δC 39.5 for DMSO-d6, δH 2.04 and δC 29.8 for acetone-d6). ESIMS spectra were measured on a Bruker amaZon speed ETD or Waters Xevo G2-XS Qtof mass spectrometer. The preparative HPLC system comprised a Waters 515 HPLC pump, Waters 2489 UV/visible detector, and Pantos Unicorder U-228.
Fungal material
The fungal strains 17F4103 and 17F4110 used in this study were isolated from soil collected in Miyazaki Prefecture, Japan, in March 2017, and deposited at the Graduate School of Pharmaceutical Sciences, Kumamoto University. The strains were identified according to their ITS sequences. A 600 base pair ITS sequence of 17F4103 had 100% sequence identity to that of Talaromyces pinophilus (CP017345), and a 624 base pair ITS sequence of 17F4110 had 99.42% sequence identity to that of Paraphaeosphaeria sp. (KM103318). The sequence data of these strains had been deposited to GenBank with accession numbers, MT093464 (17F4103), and MT093465 (17F4110).
Fermentation, extraction, and isolation
The fungi T. pinophilus (17F4103) and Paraphaeosphaeria sp. (17F4110) were co-cultured on a malt extract agar medium (2.0% malt extract, 0.5% peptone, and 1.5% agar; 40 mL) in 200 plastic square plates (140 × 100 × 14.5 mm) at 25 °C for 35 days. The culture was extracted with MeOH. The MeOH extract was concentrated and partitioned between EtOAc and H2O. The EtOAc fraction was then partitioned between n-hexane and 90% MeOH–H2O, and the H2O fraction was partitioned between n-BuOH and H2O. The n-BuOH fraction (2.5 g) was subjected to ODS chromatography with a stepwise gradient of MeOH–H2O. The fraction eluted with 40% MeOH–H2O was purified by HPLC [Asahipak GS-310P (21.5 × 500 mm), Asahi Chemical Industry Co., Ltd., 40% MeCN–H2O; COSMOSIL 5C18-MS-II column (20 × 250 mm), Nacalai Tesque Inc., 20% MeOH–H2O (0.1% acetic acid)] to afford talarodone A (1, 0.80 mg). The 90% MeOH–H2O fraction (1.1 g) was subjected to SiO2 chromatography with 5, 10, and 20% MeOH–CH2Cl2. The fraction eluted with 5% MeOH–CH2Cl2 was purified by SiO2 HPLC [Inertsil SIL-100A (20 × 250 mm), GL Sciences Inc., CH2Cl2–MeOH (319:1)] to afford 2 (6.7 mg), 3 (14.0 mg), 4 (3.0 mg), and 5 (4.5 mg). The fraction eluted with 10% MeOH–CH2Cl2 was subjected to ODS chromatography with 75% MeOH–H2O and MeOH. The fraction eluted with MeOH was purified by HPLC [Inertsil Diol (20 × 250 mm), GL Sciences Inc., CH2Cl2–MeOH (24:1)] to afford 6 (3.4 mg).
Talarodone A (1): white amorphous powder. [α]D20 − 1.8 (c = 0.33, 50% MeOH–H2O). UV λmax (50% MeOH–H2O) (log ε): 318 (3.82), 260 (3.97), 236 (4.39), 206 (4.63), 196 (4.59) nm. IR (film) υmax: 3244, 2922, 2849, 1664, 1599, 1330, 1063, 843, 791 cm−1. 1H and 13C NMR data (DMSO-d6), see Table 1. HRTOFMS m/z 406.1528 [M+H]+ (calcd for C20H24NO8, 406.1502).
Conformational analyses and chemical shift calculations for the γ-pyridone and γ-pyridol forms of 1 and 3
These experiments were performed as previously described [15] using spartan’18 instead of spartan’16. Chemical shift calculations were performed at the ωB97X-D/6-31G* level.
References
Newman DJ, Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79:629–661
Newman DJ (2016) Predominately uncultured microbes as sources of bioactive agents. Front Microbiol 7:1832
Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG, Fischbach MA (2014) Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158:412–421
Katz M, Hover BM, Brady SF (2016) Culture-independent discovery of natural products from soil metagenomes. J Ind Microbiol Biotechnol 43:129–141
Scherlach K, Hertweck C (2009) Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem 7:1753–1760
Moon K, Xu F, Seyedsayamdost MR (2019) Cebulantin, a cryptic lanthipeptide antibiotic uncovered using bioactivity-coupled HiTES. Angew Chem Int Ed 58:5973–5977
Li HT, Zhou H, Duan RT, Li HY, Tang LH, Yang XQ, Yang YB, Ding ZT (2019) Inducing secondary metabolite production by co-culture of the endophytic fungus Phoma sp. and the symbiotic fungus Armillaria sp. J Nat Prod 82:1009–1013
Bertrand S, Schumpp O, Bohni N, Monod M, Gindro K, Wolfender JL (2013) De novo production of metabolites by fungal co-culture of Trichophyton rubrum and Bionectria ochroleuca. J Nat Prod 76:1157–1165
Sadahiro Y, Kato H, Williams RM, Tsukamoto S (2020) Irpexine, a new isoindolinone alkaloid produced by co-culture of endophytic fungi, Irpex lacteus and Phaeosphaeria oryzae. submitted
Ge HM, Shen Y, Zhu CH, Tan SH, Ding H, Song YC, Tan RX (2008) Penicidones A-C, three cytotoxic alkaloidal metabolites of an endophytic Penicillium sp. Phytochemistry 69:571–576
Liu Y, Yang Q, Xia G, Huang H, Li H, Ma L, Lu Y, He L, Xia X, She Z (2015) Polyketides with α-glucosidase inhibitory activity from a mangrove endophytic fungus, Penicillium sp. HN29-3B1. J Nat Prod 78:1816–1822
Buommino E, Tirino V, De Filippis A, Silvestri F, Nicoletti R, Ciavatta ML, Pirozzi G, Tufano MA (2011) 3-O-Methylfunicone, from Penicillium pinophilum, is a selective inhibitor of breast cancer stem cells. Cell Prolif 44:401–409
Chen MJ, Fu YW, Zhou QY (2014) Penifupyrone, a new cytotoxic funicone derivative from the endophytic fungus Penicillium sp. HSZ-43. Nat Prod Res 28:1544–1548
Myobatake Y, Takeuchi T, Kuramochi K, Kuriyama I, Ishido T, Hirano K, Sugawara F, Yoshida H, Mizushina Y (2012) Pinophilins A and B, inhibitors of mammalian A-, B-, and Y-family DNA polymerases and human cancer cell proliferation. J Nat Prod 75:135–141
Torii M, Kato H, Hitora Y, Angkouw ED, Mangindaan REP, de Voogd NJ, Tsukamoto S (2017) Lamellodysidines A and B, sesquiterpenes isolated from the marine sponge Lamellodysidea herbacea. J Nat Prod 80:2536–2541
Acknowledgements
This work was supported by JSPS KAKENHI Grants 17H03994 (S.T.), 18K06719 (H.K.), and 18K14933 (Y. H.), from the Noda Institute for Scientific Research (S.T.), and Useful and Unique Natural Products for Drug Discovery and Development (UpRod), Program for Building Regional Innovation Ecosystems at Kumamoto University, Japan. We are grateful to Dr. K. Miyazaki and Dr. A. Kinoshita of Kyushu Research Center, Forestry and Forest Products Research Institute, Japan, for identification of the fungal isolates.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Murakami, S., Hayashi, N., Inomata, T. et al. Induction of secondary metabolite production by fungal co-culture of Talaromyces pinophilus and Paraphaeosphaeria sp.. J Nat Med 74, 545–549 (2020). https://doi.org/10.1007/s11418-020-01400-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-020-01400-1