
Abstract
Neuronal cell death induced by amyloid-β (Aβ) oligomers is implicated in neuronal degeneration and is a leading cause of Alzheimer’s disease (AD). Therefore, to identify effective therapeutic agents for AD, we investigated the neuroprotective effects of two naturally occurring retinoid X receptor (RXR) agonists (SPF1 and SPF2), isolated from the root of Sophora tonkinensis Gagnep., on the Aβ25–35-induced cytotoxicity against nerve growth factor-differentiated rat pheochromocytoma (PC12) cells. Pretreatment with SPFs significantly prevented Aβ25–35-induced apoptosis in PC12 cells, similarly to the synthetic RXR agonist bexarotene. These effects were blocked by the RXR antagonist PA452. When the effects of SPFs were studied in the presence of the liver X receptor (LXR) agonist T0901317, the protective effects of SPFs were enhanced, suggesting that RXR/LXR heterodimers may play a key role in the neuroprotective effects of SPFs. SPFs and T0901317 induced ATP-binding cassette transporter 1 (ABCA1) protein expression in PC12 cells when administered alone or in combination. Intriguingly, a functional inhibitor of ABCA1 cyclosporine A negated the neuroprotective effects of SPFs or T0901317. Taken together, these results demonstrate that the RXR agonists SPF1 and SPF2 protect PC12 cells from Aβ25–35-induced neurotoxicity in an RXR-dependent manner and that their effects are markedly enhanced by the LXR agonist T0901317, in part related to ABCA1 function. These results suggest a novel approach to the treatment or prevention of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and a major cause of dementia, characterized by the deposition of amyloid-β (Aβ) peptide, a principal component of senile plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein [1, 2]. Currently, the precise mechanism underlying the pathogenesis of AD has not been fully elucidated, but the predominant theory for AD, the “amyloid cascade hypothesis,” postulates that the deposition of Aβ peptides in the brain is intimately linked with neuronal degeneration leading to cognitive deficit. Aβs sequentially cleaved from amyloid precursor protein have multiple forms, some of which are highly neurotoxic and exert diverse effects on neuronal functions, finally leading to neuronal cell death. Neurotoxic oligomers such as Aβ42 oligomer interact with various receptors, including the receptor for advanced glycation end products (RAGE), NMDA-type glutamate receptor (NMDAR), calcium-sensing receptors (CaSRs), and cellular prion protein (PrPc) expressed on the surface of neurons, which trigger signals to induce inflammation, endoplasmic reticulum (ER) stress, and oxidative stress, and elevate intracellular Ca2+ levels [3, 4]. The failure of several recent clinical trials using anti-Aβ antibody to reduce Aβ deposits has provoked controversy as to whether Aβ plaque deposits are functionally associated with the pathogenesis of AD [5, 6]. However, at present, it seems likely that several interacting pathophysiological processes induced by Aβ oligomers might be intricately involved in neuronal degeneration. Given the urgent need for the development of novel and effective therapeutic strategies for the prevention and treatment of AD, it is worth exploring therapeutic agents that protect neuronal cells from Aβ oligomer-evoked neurotoxicity.
Nuclear receptors (NRs) belong to a superfamily of ligand-activated transcription factors that regulate the expression of diverse genes involved in a wide variety of biological processes, such as cellular metabolism and neuronal function, by binding to sequence-specific DNA elements. Over the past decade, numerous studies have revealed that agonists against NRs, including peroxisome proliferator-activated receptors (PPARs), liver X receptor (LXR), retinoic acid receptor, and retinoid X receptor (RXR), have a broad range of salutary effects in murine models of AD [7,8,9,10,11]. The beneficial effects of NR agonists are broadly categorized into stimulation of Aβ clearance, suppression of Aβ generation, regulation of neuronal function, and anti-inflammatory action [12]. Animal studies have reported promising results, although there have been some inconsistencies between reports [1]. Additionally, the clinical efficacy for AD treatment has not yet been proven because of low blood–brain barrier (BBB) permeability and serious adverse effects inherent in some NR agonists [13]. Overall, even though some problems remain to be addressed, the activation of NRs seems to be a potential therapeutic strategy for AD treatment.
The RXR selective agonist bexarotene (BEX) was reported to enhance soluble Aβ clearance, reduce Aβ plaque burden, and reverse cognitive deficits in murine models of AD [14]. However, the beneficial effects of BEX on Aβ metabolism and plaque burden are still controversial [15], although recent evidence demonstrated that BEX affected signaling pathways related to neuronal differentiation and neuron projections [16], and rescued the age-related decline of synaptic proteins PSD95, GluR1, and NR1 which regulate synaptic plasticity [17], suppress inflammation and astrogliosis, and activate microglial phagocytosis [8]. Taken together, BEX seems to have neuroprotective effects mediated via multifaceted mechanisms. However, serious side effects following BEX treatment have been reported, including hyperlipidemia [18], hypothyroidism [19], hepatomegaly [20], and anemia [21], which limit the usage of BEX for the treatment of cutaneous T-cell lymphoma. In this context, alternative RXR agonists with a gene expression profile, bioavailability, and BBB permeability distinct from BEX are required for AD treatment or prophylaxis.
In our previous study, two prenylated flavonoids referred to as SPF1 and SPF2 were isolated from the roots of Sophora tonkinensis Gagnep., a Chinese folk medicine. SPF1 and SPF2 showed selective RXR agonist activities with EC50 values of 0.77 μM and 0.78 μM, respectively [22]. SPFs activate PPARs/RXR and RXR/LXR heterodimers, which are classified as permissive heterodimers, alone or in combination with the ligands for PPARs or LXR. They also increase ATP-binding cassette transporter 1 (ABCA1) and apoE mRNA expression similarly to BEX. However, the gene expression profiles induced by BEX and SPF1 are not identical and only a limited number of genes overlap (unpublished data). Three isoforms of RXRs have been identified and these are implicated in the diverse regulation of gene transcription based on their ability to dimerize with various partner NRs and to form homodimers or tetramers [23]. In addition, the recruitment of various RXR ligand-dependent cofactors increases the diversity of RXR-regulated gene transcription [24]. Thus, considering the pleiotropic effects of RXR with different properties from other NRs, alternative RXR activators with neuroprotective effects, but not adverse effects, will be potential candidates for therapeutic agents to prevent or treat AD. Therefore, we investigated the neuroprotective effects of SPF1 and SPF2 and found that they protected PC12 cells from Aβ-induced neurotoxicity by a mechanism dependent on RXR/LXR heterodimers.
Materials and methods
Reagents
2-[{3-Hydroxy-2′,2-dimethyl-8-(3-methyl-2-butenyl)}chroman-6-yl]-7-hydroxy-8-(3-methyl-2-butenyl)-chroman-4-one (SPF1) and 2-[{2-(1-hydroxy-1-methylethyl)-7-(3-methyl-2-butenyl)-2′,3-dihydrobenzofuran}-5-yl]-7-hydroxy-8-(3-methyl-2-butenyl)-chroman-4-one (SPF2) were isolated from the root of Sophora tonkinensis Gagnep. as described in our previous paper [22]. The RXR agonist bexarotene (BEX) and LXR agonist T0901317 (T090) were purchased from Cayman Chemicals (Denver, CO, USA). All chemicals were dissolved in dimethyl sulfoxide (DMSO) (Fujifilm Wako Pure Chemical, Osaka, Japan) and stored at − 20 °C until use. Amyloid-β peptide (Aβ25–35) was obtained from Peptide Institute, Inc. (Osaka, Japan). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St. Louis, MO, USA). RPMI1640 medium, penicillin, streptomycin, and poly-l-lysine (PLL) were obtained from Fujifilm Wako Pure Chemical. Fetal bovine serum (FBS), horse serum (HS), and nerve growth factor (NGF) were purchased from Nichirei Biosciences Inc. (Tokyo, Japan), Gibco (Grand Island, NY, USA), and PeproTech, Inc. (Rocky Hill, NJ, USA), respectively. Cyclosporine A (CsA) was obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan).
Cell culture
The rat pheochromocytoma cell line PC12 and hepatocellular carcinoma cell line HepG2 were obtained from the JCRB Cell Bank (Osaka, Japan). PC12 cells were maintained in RPMI1640 medium supplemented with 10% HS, 5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere of 5% CO2 in air. PC12 cells were differentiated with 50 ng/ml NGF for 3 days into sympathetic neuron-like cells and the differentiated PC12 cells were then used in the following experiments. HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere of 5% CO2 in air.
Cell viability assay
Cell viability was determined by MTT assay. Briefly, PC12 cells were seeded into PLL-coated 96-well microplates at a density of 3 × 105 cells/ml. The cells were treated with SPF1 (1 µM), SPF2 (1 µM), and BEX (1 µM) for 12 h, followed by exposure to Aβ25–35 (0.5 µM). Aβ25–35 was dissolved in DMSO, diluted to 5% DMSO solution with sterile distilled H2O, and stored until use. For all cell cultures, the concentration of DMSO was less than 0.025%. After 48 h incubation, the cells were thoroughly washed twice with phosphate-buffered saline (PBS), and MTT solution was added to the culture at a final concentration of 0.5 mg/ml and incubated for another 6 h at 37 °C. Sodium dodecyl sulfate (SDS) was then added for 18 h to dissolve formazan crystals. The absorbance of each well was determined at 570 nm by microplate reader (Beckman Coulter®, Brea, CA, USA). The cell viability was calculated as the percentage of the control group.
Detection of apoptotic cells by Hoechst 33342 dye staining
PC12 cells were seeded onto PLL-coated cover glasses at a density of 5 × 104 cells/ml. After treatment with the compounds, then Aβ25–35, the cells were fixed in 4% paraformaldehyde for 20 min at room temperature and washed with PBS 3 times. Thereafter, the cells were stained with 2 μg/ml Hoechst 33342 dye for 1 h at room temperature.The cells were then thoroughly washed with PBS 3 times before observation with a BZ-9000 fluorescence microscope (Keyence Corp., USA). The dye was excited at 360 nm emission and filtered at 460 nm. Apoptotic cells were determined as cells with condensed nuclei and strong bright Hoechst 33342 staining. Total cells and apoptotic cells in 12 randomly selected fields were counted by two persons. The result was expressed as the percentage of apoptotic cells to the total cells.
Total RNA extraction and real-time qPCR
PC12 cells were seeded into 24-well PLL-coated plates at a density of 2 × 105 cells/ml. After treatment with the compounds, total RNA was extracted from cells using RNAiso Plus (Takara Bio Inc., Kusatsu, Japan) according to the manufacturer’s instructions. A total of 250 ng RNA was reverse-transcribed using ReverTra Ace (Toyobo, Japan). Quantitative real-time PCR (RT-qPCR) with SYBR green (Thermo Fisher Scientific, K.K., Tokyo, Japan) was performed using the TP800 Thermal Cycler Dice® Real-time System (Takara Bio. Inc., Shiga, Japan). qPCR consisted of the following conditions: 50 cycles of 5 s at 95 °C and 30 s at 63 °C. The relative expression of target gene mRNA was calculated by the ΔΔCt method and normalized to the amount of β-actin expression (Table 1).
Total protein extraction and Western blot analysis
PC12 cells were seeded into 6-well PLL-coated plates at a density of 2 × 105 cells/ml. After treatment with the compounds, total protein was extracted from the cells with RIPA lysis buffer, and then the protein concentration was determined by Bradford assay (Bio-Rad, USA). Protein samples were separated by SDS-PAGE and transferred to PVDF membranes (Immobilon®, USA). The membranes were blocked with 3% (w/v) BSA in TBS-T buffer (Tris-buffered saline containing 0.1% Tween 20) at room temperature for 2 h and incubated with anti-ABCA1 antibody (1:1000, Abcam) and anti-β-actin antibody (1:1000, BioLegend) at 4 °C overnight. The next day, the membranes were washed with TBS-T 3 times and incubated with horseradish peroxidase-linked anti-mouse IgG antibody (1:5000, Cell Signaling Technology) at room temperature for 1 h. The membranes were then washed with TBS-T 3 times and bands were detected by using an enhanced chemiluminescence reagent (ImmunoStar®, Fujifilm Wako Pure Chemical), and imaged by ImageQuant LAS4000mini (GE Healthcare, USA).
Statistical analysis
Data are presented as the mean ± SD. The significance of differences was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. p values less than 0.05 were considered statistically significant.
Results
Effect of SPFs on Aβ25–35-induced cytotoxicity in PC12 cells
To explore the neuroprotective effects of the naturally occurring RXR agonists shown in Fig. 1 on Aβ25–35-induced neuronal cell death, SPF1, SPF2, and BEX were incubated with NGF-differentiated PC12 cells with characteristics of sympathetic neuron-like cells and cell viability was evaluated using the MTT method (Fig. 2). In this experiment, the concentration of Aβ25–35 peptide was set at 0.5 µM, where about 50% of cells were killed according to the results of a preliminary experiment. The concentration of SPFs was also set at 1 µM, where SPFs had the most potent effects in preliminary experiments. SPF1 and SPF2 exhibited no cytotoxicity against PC12 cells, similarly to BEX. Following a 48-h incubation with Aβ25–35 at a concentration of 0.5 μM, cell viability was reduced to approximately 50% of the control group, while SPFs and BEX pretreatment for 12 h significantly reduced Aβ25–35-induced cytotoxicity in PC12 cells.

Chemical structures of SPF1, SPF2, and bexarotene

Effect of SPFs on the cell viability of Aβ25–35-treated PC12 cells. NGF-differentiated PC12 cells were treated with 1 µM SPF1, SPF2, or BEX for 12 h, followed by 48-h exposure to 0.5 μM Aβ25–35. Cell viability was measured by the MTT method. Data represent the mean ± SD of six determinants of a representative experiment from two independent experiments with similar results. Statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. p values less than 0.05 were considered significant. **p < 0.01 vs. Aβ25–35-treated vehicle group (Veh)
Effect of SPFs on Aβ25–35-induced apoptosis in PC12 cells
To further study the protective effect of SPFs on Aβ25–35-induced cell death, PC12 cells were stained with Hoechst 33342 to assess the characteristics of pyknotic nuclei in apoptotic cells as previously reported [25]. Aβ25–35 treatment increased the percentage of apoptotic cells with cell shrinkage, chromatin condensation, and strong bright fluorescent nuclei, whereas SPFs or BEX reduced the apoptotic cell population in PC12 cells treated with Aβ25–35 (Fig. 3a, b).

Protective effects of SPFs on Aβ25–35-induced apoptosis in PC12 cells. NGF-differentiated PC12 cells were treated with 1 µM SPF1, SPF2, or BEX for 12 h, followed by 48-h exposure to 0.5 µM Aβ25–35. Cells were stained with Hoechst 33342 and then apoptotic cells with pyknotic nuclei were examined under a fluorescence microscope as described in Materials and Methods (Fig. 3a). Data represent the mean ± SD of three wells of a representative experiment from two independent experiments with similar results. The y-axis indicates the percentage of apoptotic cells to total cells (Fig. 3b). Statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. p values less than 0.05 were considered significant. **p < 0.01 vs. Aβ25–35-treated vehicle group (Veh)
Involvement of RXR in the protective effects of SPFs on Aβ25–35-induced apoptosis
To investigate whether RXR is involved in the protective effect of SPFs, the RXR-specific pan-antagonist PA452 was used. SPFs significantly inhibited Aβ25–35-induced apoptosis in PC12 cells, and the inhibitory effects were reversed by PA452 treatment (Fig. 4). BEX also exerted an inhibitory effect on Aβ25–35-induced apoptosis, and its effect was suppressed by PA452. Similar results were obtained when another RXR antagonist, HX531, was used instead of PA452 (data not shown).

Effects of RXR antagonist on protective effect of SPFs on Aβ25–35-induced apoptosis in PC12 cells. NGF-differentiated PC12 cells were treated with 1 µM SPF1, SPF2, or BEX for 12 h in the presence or absence of the RXR antagonist PA452 (1 μM), followed by 48-h exposure to 0.5 μM Aβ25–35. Data represent the mean ± SD of three wells of a representative experiment from two independent experiments with similar results. Statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. p values less than 0.05 were considered significant. **p < 0.01 vs. Aβ25–35-treated vehicle group (Veh), ##p < 0.01 between the indicated groups in the presence or absence of PA452
Effect of SPFs in combination with an LXR agonist on Aβ25–35-induced apoptosis
In a previous study, we demonstrated that SPFs activated RXR/LXR heterodimers more effectively than PPARγ/RXR heterodimers in the presence of an agonist for partner NR [22]. Therefore, we examined the effects of SPFs in combination with the LXR agonist T0901317 on Aβ25–35-induced apoptosis in PC12 cells. Interestingly, T0901317 alone had a protective effect on Aβ25–35-induced apoptosis in PC12 cells, and the effects of SPFs were significantly potentiated in the presence of T0901317 (Fig. 5).

Protective effects of SPFs in combination with T0901317 on Aβ25–35-induced apoptosis in PC12 cells. NGF-differentiated PC12 cells were treated with 1 µM SPF1, SPF2, or BEX for 12 h in the presence or absence of the LXR agonist T0901317 (T090) (1 μM), followed by 48-h exposure to 0.5 μM Aβ25–35. Data represent the mean ± SD of three wells. Statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. p values less than 0.05 were considered significant. **p < 0.01 vs. Aβ25–35-treated vehicle group (Veh), ##p < 0.01 between the indicated groups in the presence or absence of T0901317
Involvement of ABCA1 in the protective effects of SPFs and LXR agonist on Aβ25–35-induced apoptosis in PC12 cells
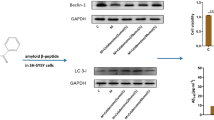
ATP-binding cassette transporter A1 (ABCA1), a well-known target gene regulated by LXR and RXR, is involved in cholesterol efflux for HDL generation or the regulation of signal transduction by modulating plasma membrane lipid composition [26]. Therefore, the mRNA and protein levels of ABCA1 were determined after treating PC12 cells for 24 h with SPFs at a concentration of 1 μM. As shown in Fig. 6, SPFs induced ABCA1 mRNA and protein expression, and their induction was potentiated by the LXR agonist T0901317. Similar results were obtained with BEX. These results indicated that SPFs exert synergistic effects on ABCA1 expression via the RXR/LXR heterodimer. To further elucidate whether the protective effects of SPFs were associated with ABCA1 function, cyclosporine A (CsA), which inhibits the function of ABCA1 cholesterol efflux by blocking ABCA1 and ApoA-1 binding ability [27], was used. As shown in Fig. 7, CsA partially, but significantly, reversed the protective effects of SPFs on Aβ25–35-induced apoptosis in PC12 cells, as well as those of BEX and T0901317. These results suggest that the protective effects of SPFs on Aβ25–35-induced apoptosis in PC12 cells are partially dependent on ABCA1 function.

ABCA1 a mRNA or b protein levels after treatment of PC12 cells with SPFs and BEX in the presence or absence of T0901317. NGF-differentiated PC12 cells were treated with 1 µM SPF1, SPF2, or BEX for 24 h in the presence or absence of T0901317 (T090) (0.1 μM). The mRNA and protein levels of ABCA1 were assessed using RT-qPCR and Western blotting, respectively. mRNA levels were normalized relative to β-actin mRNA levels and then represented as a fold change from the control group (Ctrl). The data represent the mean ± SD of a representative experiment from two independent experiments with similar results, and were evaluated for statistical significance by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. *p < 0.05, **p < 0.01 vs. a T09013170treated vehicle group (Veh) (n = 3), b vehicle group (Ctrl) or T0901317-treated vehicle group (Veh) (n = 3)

Effects of cyclosporine A on protective effect of SPFs on Aβ25–35-induced apoptosis in PC12 cells. NGF-differentiated PC12 cells were treated with 1 µM SPF1, SPF2, or BEX for 12 h in the presence or absence of cyclosporine A (CsA) (2.5 μM), followed by 48-h exposure to 0.5 μM Aβ25–35. Data represent the mean ± SD of three wells of a representative experiment from two independent experiments with similar results. Statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. p values less than 0.05 were considered significant. **p < 0.01 vs. Aβ25–35-treated vehicle group (Veh), #p < 0.05, ##p < 0.01 between the indicated groups in the presence or absence of CsA treatment
Effects of SPFs and BEX on sterol regulatory element-binding protein 1c (SREBP-1c) mRNA expression in HepG2 cells
BEX causes some serious side effects [18,19,20,21]. Therefore, we used HepG2 cells to determine whether SPFs induced the mRNA expression of SREBP-1c, which is considered a causal gene for hyperlipidemia. BEX and the LXR agonist T0901317 significantly increased the mRNA levels of SREBP-1c, whereas SPFs failed to show such an effect (Fig. 8).

Effects of SPFs and BEX on SREBP-1c mRNA expression in HepG2 cells. HepG2 cells were treated with 10 µM SPF1, SPF2, BEX, or T0901317 for 12 h. The mRNA levels of SREBP-1c were assessed using RT-qPCR. mRNA levels were normalized relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels and then represented as a fold change from the control group (Ctrl). The data represent the mean ± SD, and were evaluated for statistical significance by one-way analysis of variance (ANOVA) followed by Bonferroni’s t test. *p < 0.05, **p < 0.01 vs. vehicle group (Ctrl)
Discussion
Accumulating evidence has demonstrated that Aβ, especially Aβ-oligomers, transmit or block a variety of signals to neuronal cells via various receptors or channels, finally resulting in neuronal cell death [3]. Given the urgent need for the development of effective therapeutic agents for AD, we focused on Aβ oligomer-induced neurotoxicity as a therapeutic target and sought to evaluate the potential of RXR agonist SPFs. We used NGF-differentiated PC12 cells as an in vitro model to study Aβ oligomer-induced neurotoxicity. We used Aβ25–35 instead of Aβ1–40 or Aβ1–42 because the Aβ25–35 fragment is a functional neurotoxic domain that is detected in AD patients [28, 29]. In this study, the pretreatment of PC12 cells with SPFs effectively reduced cell death (apoptosis) induced by Aβ25–35 and the RXR antagonist PA452 abolished the protective effects of SPFs. The synthetic RXR agonist BEX also had a similar effect on Aβ25–35-induced apoptosis, which indicated that SPFs exert protective effects on Aβ25–35-induced neurotoxicity via RXR. On the basis of our previous study showing that SPFs activated RXR/LXR heterodimers more effectively than PPARγ/RXR heterodimers [22], the impact of the LXR agonist T0901317 on Aβ25–35-induced apoptosis was studied. T0901317 alone decreased Aβ25–35-induced apoptosis and potentiated the anti-apoptotic effect in the presence of SPFs. These results strongly suggest that SPFs activate RXR/LXR heterodimers to protect PC12 cells from Aβ25–35-induced neurotoxicity, and the protective effect was enhanced in combination with T0901317. To examine the mechanism underlying the protective effect on Aβ25–35-induced neurotoxicity, the expressions of ABCA1 mRNA and protein were assessed by RT-qPCR and Western blot analysis, respectively. ABCA1 is transcriptionally regulated by LXR and RXR, and a loss-of-function mutation in ABCA1 or polymorphisms in ABCA1 are associated with a high risk of AD [30, 31], which suggests that ABCA1 has an important role in protection against AD development and the protection of neuronal cells from Aβ25–35-induced neurotoxicity. Treatment of PC12 cells with SPFs alone increased ABCA1 expression and this was synergistically increased in the presence of T0901317. Intriguingly, the ABCA1 protein levels correlated with the degree of the SPFs’ protective effect with or without T0901317. Therefore, CsA, a functional inhibitor of ABCA1, was added to PC12 cells, followed by Aβ25–35 treatment. CsA significantly reduced the protective effects of SPFs, BEX, and T0901317 on Aβ25–35-induced neurotoxicity, although CsA alone did not affect Aβ25–35-induced apoptosis. Thus, SPFs might exert protective effects on Aβ25–35-induced neurotoxicity by inducing the production of ABCA1 protein via RXR/LXR heterodimers.
ABCA1 plays critical roles not only in cholesterol efflux to lipid-free apoA1 and apoE for HDL generation, but also in the regulation of plasma membrane lipid composition, especially the decrease of cholesterol level in lipid rafts, specialized membrane lipid domains implicated in signal transduction, neurotransmission, and ligand–receptor interaction [26]. T0901317 and platycodin D were reported to inhibit lipopolysaccharide-induced inflammatory responses in mammary or lung epithelial cells by activating the LXR–ABCA1 signaling pathway to reduce the cholesterol levels of lipid rafts and the translocation of Toll-like receptor 4 (TLR4) into the lipid rafts [32, 33]. Aβ triggers signals to induce inflammation, ER stress, oxidative stress, and elevated levels of intracellular Ca2+ [3, 4], which cause neuronal cell death. Many potential Aβ-oligomer binding proteins or receptors have been proposed to date, including RAGE, NMDAR, CaSRs, and TLR4, which are most likely to evoke inflammatory-related signals in neuronal cells [34,35,36,37]. In particular, RAGE is expressed on neurons, and is markedly increased in AD [38]. Furthermore, the activation of RAGE by Aβ generates reactive oxygen species (ROS) and induces inflammatory signals via mitogen-activated protein kinase or nuclear factor-κB activation, finally resulting in neuronal cell death [39]. However, whether ABCA1 is implicated in the regulation of RAGE function by altering cholesterol levels in plasma membranes remains to be determined to further elucidate the effects of SPFs. In addition, Aβ activates TLR4 expressed on neurons to induce inflammatory signals. TLR4 is widely expressed on microglia, astrocytes, and neurons in rodents, but not on neurons in humans [40]. In this study, PC12 cells derived from rat pheochromocytoma were used, suggesting that the effects of SPFs might be explained by the modulation of TLR4 function by increased ABCA1. However, the species difference in the expression of TLR4 among cell types indicates the necessity of more detailed studies to evaluate the efficacy of SPFs in AD patients.
Numerous naturally occurring plant-derived products have been reported to protect neuronal cells from Aβ oligomer-induced neurotoxicity via diverse mechanisms. For example, cyanidin suppressed Aβ oligomer-induced inflammation and ROS production via the TLR4/NADPH oxidase pathway [41, 42]. Epigallocatechin gallate (EGCG) inhibited ER stress [43], hesperidin reversed Aβ-induced mitochondrial dysfunction and ROS production [44], and icariin attenuated tau protein hyperphosphorylation [45]. Thus, there are many naturally occurring products with the potential to treat AD patients, although further detailed studies are required to elucidate their efficacy in vivo or in clinical studies.
In the present study, we demonstrated that SPFs protected PC12 cells from Aß25–35-induced neurotoxicity in a similar manner to BEX. However, BEX or T0901317, but not SPFs, increased the mRNA levels of SREBP-1c, a causal gene for hyperlipidemia, in HepG2 cells, the most common side effect of BEX or LXR agonists. Thus, SPFs might be RXR agonists that induce fewer side effects compared with BEX.
In conclusion, we demonstrated that the RXR agonists SPF1 and SPF2 protected PC12 cells from Aβ25–35-induced neurotoxicity in an RXR-dependent manner, that their effects were markedly enhanced by the LXR agonist T0901317, and were in part related to ABCA1 function. Thus, it becomes evident that SPF1 and SPF2 possess properties distinct from other naturally occurring products reported to have beneficial effects on neurotoxicity. Therefore, the results of this study may provide a novel approach to the future treatment or prevention of AD. However, the precise mechanism involved and their efficacy in vivo using model animals remain to be determined.
Change history
09 April 2019
In the original publication of the article, Table 1 and Fig. 1 were incorrectly published.
References
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6:487–498
Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM (2016) Amyloid-β receptors: the good, the bad, and the prion protein. J Biol Chem 291:3174–3183
Chiarini A, Armato U, Liu D, Dal Pra I (2016) Calcium-sensing receptors of human neural cells play crucial roles in Alzheimer’s disease. Front Physiol 7:134
Mullard A (2016) Alzheimer amyloid hypothesis lives on. Nat Rev Drug Discov 16:3–5
Herrup K (2015) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18:794–799
Inestrosa NC, Godoy JA, Quinatanilla RA, Koenig CS, Bronfman M (2005) Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents beta-amyloid neurodegeneration: role of Wnt signaling. Exp Cell Res 304:91–104
Mariani MM, Malm T, Lamb R, Jay TR, Neilson L, Casali B, Medarametla L, Landreth GE (2017) Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci Rep 7:42270
Malm T, Mariani MM, Donovan LJ, Neilson L, Landreth GE (2015) Activation of the nuclear receptor PPARδ is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. J Neuroinflamm 12:7
Katsuki H, Kurimoto E, Takemori S, Kurauchi Y, Hisatsune A, Isohama Y, Izumi Y, Kume T, Shudo K, Akaike A (2009) Retinoic acid receptor stimulation protects midbrain dopaminergic neurons from inflammatory degeneration via BDNF-mediated signaling. J Neurochem 110:707–718
Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P (2007) Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver X receptors. Proc Natl Acad Sci USA 104:10601–10606
Moutinho M, Landreth GE (2017) Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J Lipid Res 58:1937–1949
Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, Craft S, Landreth G, Linnamägi U, Sawchak S (2010) Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 30:131–146
Cramer PE, Cirrito JR, Wesson DW, Lee CYD, Kario JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE (2012) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335:1503–1506
Landreth GE, Cramer PE, Lakner MM, Cirrito JR, Wesson DW, Brunden KR, Wilson DA (2013) Response to comments on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340:924
Mounier A, Georgiev D, Nam KN, Fitz NF, Castranio EL, Wolfe CM, Cronican AA, Schug J, Lefterov I, Koldamova R (2015) Bexarotene-activated retinoid X receptors regulate neuronal differentiation and dendritic complexity. J Neurosci 35:11862–11876
Tachibana M, Shinohara M, Yamazaki Y, Liu CC, Rogers J, Bu G, Kanekiyo T (2016) Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Exp Neurol 277:1–9
Pinaire JA, Reifel-Miller A (2007) Therapeutic potential of retinoid X receptor modulators for the treatment of the metabolic syndrome. PPAR Res 2007:94156
Liu S, Ogilvie KM, Klausing K, Lawson MA, Jolley D, Li D, Bilakovics J, Pascual B, Hein N, Urcan M, Leibowitz MD (2002) Mechanism of selective retinoid X receptor agonist-induced hypothyroidism in the rat. Endocrinology 143:2880–2885
Lenhard JM, Lancaster ME, Paulik MA, Weiel JE, Binz JG, Sundseth SS, Gaskill BA, Lightfoot RM, Brown HR (1999) The RXR agonist LG100268 causes hepatomegaly improves glycaemic control and decreases cardiovascular risk and cachexia in diabetic mice suffering from pancreatic beta-cell dysfunction. Diabetologia 42:545–554
Al Mamun Bhuyan A, Bissinger R, Cao H, Lang F (2016) Triggering of suicidal erythrocyte death by bexarotene. Cell Physiol Biochem 40:1239–1251
Inoue M, Tanabe H, Nakashima K, Ishida Y, Kotani H (2014) Rexinoids isolated from Sophora tonkinensis with a gene expression profile distinct from the synthetic rexinoid bexarotene. J Nat Prod 77:1670–1677
Lefebvre P, Benomar Y, Staels B (2010) Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab 21:676–683
Boerma LJ, Xia G, Qui C, Cox BD, Chalmers MJ, Smith CD, Lobo-Ruppert S, Griffin PR, Muccio DD, Renfrow MB (2014) Defining the communication between agonist and coactivator binding in the retinoid X receptor α ligand binding domain. J Biol Chem 289:814–826
Gschwind M, Huber G (1995) Apoptotic cell death induced by beta-amyloid 1–42 peptide is cell type dependent. J Neurochem 65:292–300
Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS (2010) Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 51:3196–3206
Nagao K, Maeda M, Mañucat NB, Ueda K (2013) Cyclosporine A and PSC833 inhibit ABCA1 function via direct binding. Biochim Biophys Acta 1831:398–406
Zameer A, Schulz P, Wang MS, Sierks MR (2006) Single chain Fv antibodies against the 25–35 Aβ fragment inhibit aggregation and toxicity of Aβ42. Biochemistry 45:11532–11539
Kubo T, Nishimura S, Kumagae Y, Kaneko I (2002) In vivo conversion of racemized beta-amyloid ([D-Ser26]Aβ1–40) to truncated and toxic fragments ([D-Ser26]Aβ25–35/40) and fragment presence in the brains of Alzheimer’s patients. J Neurosci Res 70:474–483
Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R (2015) Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement 11:1430–1438
Chen Q, Liang B, Wang Z, Cheng X, Huang Y, Liu Y, Huang Z (2016) Influence of four polymorphisms in ABCA1 and PTGS2 genes on risk of Alzheimer’s disease: a meta-analysis. Neurol Sci 37:1209–1220
Hu X, Fu Y, Lu X, Zhang Z, Zhang W, Cao Y, Zhang N (2017) Protective effects of platycodin D on lipopolysaccharide-induced acute lung injury by activating LXRα-ABCA1 signaling pathway. Front Immunol 7:644
Wang J, Xiao C, Wei Z, Wang Y, Zhang X, Fu Y (2018) Activation of liver X receptors inhibit LPS-induced inflammatory response in primary bovine mammary epithelial cells. Vet Immunol Immunopathol 197:87–92
Chung J, An SH, Kang SW, Kwon K (2016) Ursodeoxycholic acid (UDCA) exerts anti-atherogenic effects by inhibiting RAGE signaling in diabetic atherosclerosis. PLoS One 11:e0147839
Zheng S, Yang H, Chen Z, Zheng C, Lei C, Lei B (2015) Activation of liver X receptor protects inner retinal damage induced by N-methyl-d-aspartate. Invest Ophthalmol Vis Sci 56:1168–1180
Cui HL, Guo B, Scicluna B, Coleman BM, Lawson VA, Ellett L, Meikle PJ, Bukrinsky M, Mukhamedova N, Sviridov D, Hill AF (2014) Prion infection impairs cholesterol metabolism in neuronal cells. J Biol Chem 289:789–802
Wang WY, Tan MS, Yu JT, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3:136
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Sturchler E, Galichet A, Weibel M, Leclerc E, Heizmann CW (2008) Site-specific blockade of RAGE-Vd prevents amyloid-β oligomer neurotoxicity. J Neurosci 28:5149–5158
Carty M, Bowie AG (2011) Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol 81:825–837
Thummayot S, Tocharus C, Jumnongprakhon P, Suksamram A, Tocharus J (2018) Cyanidin attenuates Aβ25–35-induced neuroinflammation by suppressing NF-κB activity downstream of TLR4/NOX4 in human neuroblastoma cells. Acta Pharmacol Sin 2018:1439–1452
Wang Y, Fu XT, Li DW, Wang K, Wang XZ, Li Y, Sun BL, Yang XY, Zheng ZC, Cho NC (2016) Cyanidin suppresses amyloid beta-induced neurotoxicity by inhibiting reactive oxygen species-mediated DNA damage and apoptosis in PC12 cells. Neural Regen Res 11:795–800
Du K, Liu M, Zhong X, Yao W, Xiao Q, Wen Q, Yang B, Wei M (2018) Epigallocatechin gallate reduces amyloid β-induced neurotoxicity via inhibiting endoplasmic reticulum stress-mediated apoptosis. Mol Nutr Food Res 62:e1700890
Wang DM, Li SQ, Zhu XY, Wang Y, Wu WL, Zhang XJ (2013) Protective effects of hesperidin against amyloid-β (Aβ) induced neurotoxicity through the voltage dependent anion channel 1 (VDAC1)-mediated mitochondrial apoptotic pathway in PC12 cells. Neurochem Res 38:1034–1044
Zeng KW, Ko H, Yang HO, Wang XM (2010) Icariin attenuates β-amyloid-induced neurotoxicity by inhibition of tau protein hyperphosphorylation in PC12 cells. Neuropharmacology 59:542–550
Acknowledgements
This work was supported by JSPS KAKENHI Grant number JP17K08352. We thank Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, W., Nakashima, Ki., Hirai, T. et al. Neuroprotective effect of naturally occurring RXR agonists isolated from Sophora tonkinensis Gagnep. on amyloid-β-induced cytotoxicity in PC12 cells. J Nat Med 73, 154–162 (2019). https://doi.org/10.1007/s11418-018-1257-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-018-1257-z