
Abstract
Phlorotannins have been reported to demonstrate several biological properties, including antioxidant activity, and activities useful in the treatment of diabetic complications and in chemoprevention of several vascular diseases. In this study, we focused on the apoptosis induced by dieckol, a marine algal phlorotannin isolated from Ecklonia stolonifera, on human hepatocellular carcinoma (HCC) Hep3B cells. Dieckol reduced the numbers of viable cells and increased the numbers of apoptotic cells in a dose-dependent manner. Immunoblotting analysis revealed that dieckol increased the expression levels of cleaved caspases-3, 7, 8, and 9, and cleaved poly(ADP-ribose) polymerase. Dieckol increased the permeability of mitochondrial membranes and the release of cytochrome c from mitochondria into the cytosol with apoptosis-inducing factor. In addition, dieckol induced increased expression of truncated Bid and Bim. The results indicate that dieckol induces apoptosis via the activation of both death receptor and mitochondrial-dependent pathways in HCC Hep3B cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer death, with an estimated worldwide incidence of over 1 × 106 new cases per year [1]. In addition, HCC is widely prevalent, especially in Asia and Africa [2]. Despite extensive searches for novel anticancer drugs and therapeutic strategies, there has been little success in improving the treatment of HCC. Only surgery offers a cure, but tumor resection is feasible for less 15 % of patients, and recurrence rates remain as high as 50 % after tumor resection due to the aggressive features of HCC, including rapid growth, resistance to chemotherapy, and the lack of effective adjunct therapy after surgery [3–6]. No effective treatment is available for this carcinoma, which has an important role in etiology related to hepatitis B and C [7]. We investigated the effects of dieckol against HCC.
Brown algae are an ingredient in many popular foods, and people ingest it as a health food in Korea and Japan. Although many studies have been performed on polyphenolic antioxidants derived from terrestrial plants, very limited information is available concerning such compounds from marine plants [8]. Marine algal polyphenols and phlorotannins, which have only been characterized in brown algae, are restricted to polymers of phloroglucinol (1,3,5-trihydroxybenzene) [9]. In previous studies, extracts of several Ecklonia species containing abundant phlorotannins were reported to possess a number of important biological properties, such as antiplasmin-inhibiting activity, HIV-1 reverse transcriptase activity, protease-inhibiting activity, antioxidation, antidiabetic complications, antiamnesia, and tyrosinase inhibitory activity [10–16]. In addition, the phlorotannins isolated from E. cava inhibited phosphorylation of JNK and p38 MAPK in human osteosarcoma cells [17]. It has been reported that the isolated eckol and dieckol from E. stolonifera inhibited both NF-kB and MMP-1 expression in human dermal fibroblasts and demonstrated possible efficacy in the prevention and treatment of skin aging [18].
Recently, several phlorotannins have been isolated from brown macroalgae [10]. We also previously reported that the ethyl acetate-soluble fraction of E. stolonifera led to the isolation of three types of phlorotannins [8]. Therefore, in this study, we focused on the apoptosis-inducing capability of dieckol, among those phlorotannins isolated from Ecklonia stolonifera. The structure of dieckol identified by comparison with published spectral data [19] is shown in Fig. 1.

Structure of dieckol isolated from E. stolonifera
Apoptosis is a major mechanism of cell death, characterized by a series of tightly regulated processes involved in the activation of a cascade of molecular events leading to cell death. Cells undergoing apoptosis are found to have elevated levels of cytochrome c in the cytosol and a corresponding decrease in the mitochondria [20]. After the release of mitochondrial cytochrome c, caspase-3 is activated [21], thereby becoming responsible for the proteolytic degradation of poly(ADP-ribose) polymerase (PARP), which occurs at the onset of apoptosis [22, 23]. In this study, we investigated the effects of dieckol on the induction of apoptosis and cytotoxicity in the HCC cell line, Hep3B. Many studies have reported that loss of control of apoptosis results in initiation and progression of cancer [24–26]. However, there is no research on dieckol-induced cell death in any cancer cell line. Thus, we studied the effects of dieckol on HCC to investigate its pathways to cell death.
This study was conducted to determine whether dieckol induces apoptosis in HCC cells and to determine the mechanisms underlying this effect. We showed that dieckol induces apoptosis in Hep3B cells via the activation of both the extrinsic/death receptor pathways and the intrinsic/mitochondrial pathway.
Materials and methods
Plant material
E. stolonifera was collected along the coast of Busan, Korea, in August 2007. The samples were rinsed using tap water to remove salt. Samples were air-dried under shade for 2 weeks and ground with a hammer grinder, and the dried powder was stored at room temperature until use.
Reagents and antibodies
All solvents were of high-performance liquid chromatography (HPLC) grade and purchased from Fisher Scientific (Pittsburgh, PA, USA). Benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (Z-VAD-fmk) was purchased from TOCRIS Bioscience (Ellisville, MO, USA). Dimethyl sulfoxide (DMSO), bovine serum albumin (BSA) and propidium iodide were obtained from Sigma Aldrich (St. Louis, MO, USA). EZ-Cytox Cell Viability Assay Solution WST-1® was purchased from Daeil Lab Service (Seoul, Korea). 4,6-Diamidino-2-phenylindole dihydrochloride hydrate (DAPI) was obtained from Roche (Mannheim, Germany). Lysis buffer consisting of 50 mM Tris–Cl (pH 7.5), 150 mM NaCl, 1 mM DTT, 0.5 % NP-40, 1 % Triton X-100, 1 % deoxycholate, 0.1 % SDS, and proteinase inhibitors (PMSF, EDTA, aprotinin, leupeptin, and prostatin A) were obtained from Intron Biotechnology (Gyeonggi, Korea). The Protein Quantification Kit (CBB solution®) was purchased from Dojindo Molecular Technologies (Rockville, MD, USA). Nitrocellulose membranes were purchased from PALL Life Sciences (Ann Arbor, MI, USA). Enhanced chemiluminescent (ECL) detection solutions were obtained from Pierce (Rockford, IL, USA). Antibodies against Bim and apoptosis-inducing factor (AIF) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Cytochrome c, Bid, Bak, β-actin, cleaved caspase-8, caspase-12, cleaved caspase-3, cleaved caspase-7, cleaved caspase-9, caspase-9, cleaved PARP, HRP conjugated anti-rabbit and anti-mouse antibodies were purchased from Cell Signaling Technology Inc. (Danvers, MA, USA).
Extraction and isolation of phlorotannins
The dried powder (4 kg) of E. stolonifera was refluxed with MeOH (3 × 9 L) for 3 h. The extract (1.1 kg) was suspended in water (1 L) and partitioned with dichloromethane, ethyl acetate (EtOAc), n-butanol, and solvents in sequence, yielding the dichloromethane (114.8 g), EtOAc (314.7 g), n-butanol (141.5 g), and water (528.2 g) fractions. The EtOAc fraction, which exhibited the most potent antioxidant activity on DPPH radical scavenging activity, was dissolved in dichloromethane and applied to a silica gel (70-230 mesh, Merck) column (100 cm × 10 cm) and eluted with a stepwise mixture of dichloromethane and methanol (6:1, 5:2, 4:3, 3:4, 2:5, 1:6, v/v, each 1.2 L). The eluates were pooled into 17 fractions based on silica gel thin-layer chromatography (TLC) (250 μm, silica gel GF Uniplate, Analtech Inc., Newark, DE, USA). The TLC plates were developed in a dichloromethane/methanol/water (65:35:10, v/v/v) solvent system. Fractions 7–9 showed high antioxidant activity on DPPH radical scavenging activities, were pooled, and were dried (25 g). The dried sample was dissolved in dichloromethane and applied to the second silica gel column (100 cm × 5 cm) to enhance the antioxidant activity. The column was eluted with a mixture of dichloromethane and methanol (6:1, 5:2, 4:3, 3:4, 2:5, 1:6, v/v, each 0.5 L) and separated into nine subfractions (Fr.1–Fr.9). Fraction 4 (5.80 g), which showed the highest antioxidant activity, was subjected to a preparative size exclusion column of Asahipak GS-310 (500 mm × 20 mm, Showa Denko, Tokyo, Japan). The exclusion HPLC apparatus consisted of a pump (Shimadzu LC-6AD), a photodiode array detector (Shimadzu SPDM20A), an online degasser (Shimadzu DUG-20A3), an autosampler (SIL-20A), a fraction collector (Shimadzu FRC-10A), a system controller (CBM-20A), and a Shimadzu LCsolution (ver. 1.22sp). Fraction 4 was chromatographed on an Asahipak GS-310 column eluting with methanol at a flow rate of 5.0 mL/min and monitored at 245 nm. The fraction was separated into five fractions (GS1–GS5). The GS3 fraction (1.110 g) showing high antioxidant activity was chromatographed over Shim-pack PREP-ODS (5 μm, 100 Å, 250 mm × 20 mm, Shimadzu Co., Tokyo, Japan). The preparative ODS HPLC system was similar to the exclusion HPLC system except for a binary pump (Shimadzu LC-6AD) and a column oven (35 °C, Shimadzu CTO-20A). The separation of the GS3 fraction was conducted using a mobile phase of 0.1 % formic acid in water (solvent A) and 0.1 % formic acid in acetonitrile (solvent B). The elution profile consisted of a linear gradient from 20 to 100 % B solvent for 40 min and re-equilibration of the column with 20 % B solvent for 10 min. The flow rate was 7.0 mL/min, and detection was performed at 245 nm. The fraction gave eight subfractions (GS3-ODS1–GS3-ODS8). GS3-ODS1 (67 mg), ODS-3 (150 mg), and ODS7 (144 mg) were purified by the same HPLC system with a Luna RP-18 column [Luna C18 (2), 5 μm, 250 mm × 10 mm, Phenomenex] and with the same mobile phase systems at a flow rate of 3.0 mL/min. The isolated fraction GS-ODS3 (dieckol) was used in this study.
Spectrometry
1H and 13C NMR spectra were determined on a JNM ECP-400 spectrometer (JEOL, Japan), using DMSO-d6 with tetramethylsilane (TMS) as an internal standard. Heteronuclear multiple quantum correlation (HMQC) and heteronuclear multiple bond correlation (HMBC) spectra were recorded using pulsed field gradients. The spectral data are as follows and the structure is shown in Fig. 1.
Structural elucidation of isolated phlorotannins
Dieckol. C36H22O18 (MW = 742). 1H NMR (400 MHz, CD3OD) δ: 6.15 (1H, s, H-300), 6.13 (1H, s, H-3), 6.09 (2H, s, H-200, 600), 6.06 (1H, d, J = 2.9 Hz, H-8), 6.05 (1H, d, J = 2.9 Hz, H-600), 5.98 (1H, d, J = 2.8 Hz, H-6), 5.95 (1H, d, J = 2.8 Hz, H-6), 5.92 (3H, s, H-20, 40, 60). 13C NMR (100 MHz, CD3OD) δ: 162.7 (C-10), 161.0 (C-30, 50), 158.6 (C-1000), 156.8 (C-7), 155.3 (C-700), 153.2 (C-3000, 50 00), 148.1 (C-200), 148.01 (C-2), 147.9 (C-900), 147.7 (C-9), 145.1 (C-5a00), 145.0 (C-5a), 144.2 (C-40 0), 144.1 (C-4000), 139.4 (C-10a), 139.3 (C-10a00), 127.3 (C-400 0), 127.0 (C-9a), 126.5 (C-1), 126.4 (C-100), 125.7 (C-9a0 0), 125.5 (C-4a00), 125.4 (C-4a), 100.7 (C-800), 100.6 (C-8), 100.3 (C-3), 100.2 (C-300), 98.5 (C-40), 97.0 (C-2000, 60 00), 96.7 (C-600), 96.6 (C-60), 96.2 (C-20, 60).
Cell culture
Hepatocellular carcinoma (Hep3B, Sk-Hep1) cells and non-cancerous human embryonic kidney (HEK293) cells were purchased from the American Tissue Culture Collection (ATCC, Manassas, VA, USA). All of the cells were cultured in Minimum Essential Medium with Earle’s Balanced Salts (MEM/EBSS) (HyClone, Logan, UT, USA), containing 10 % heat-inactivated fetal bovine serum (HyClone), 100 U/mL penicillin, and 100 μg/mL streptomycin (Cellgro Mediatech, Manassas, VA, USA) at 37 °C in a humidified atmosphere of 5 % CO2.
Cell viability assay
Exponential phases of Hep3B, Sk-Hep1 and HEK293 cells at a density of 1 × 104 cells/mL were resuspended in 100 μL of MEM medium and seeded onto 96-well plates in triplicate. Following overnight incubation, they were treated with various concentrations (70, 80, 90, 100, and 110 μM) of dieckol. The cells were incubated for 24 h, and 10 μL of WST-1® solution was added and incubated for an additional 3 h. The absorbance of the reaction was measured using an ELISA reader (Molecular Devices, Sunnyvale, CA, USA) at 460 nm and inhibitory rates were calculated. The mid-log phases of Hep3B cells were divided into four groups to compare caspase-dependent and caspase-independent cell death as follows: non-treated, Z-VAD-fmk (caspase inhibitor), dieckol, and dieckol with Z-VAD-fmk groups. After 24 h, the cells were transferred to fresh medium containing no agent (control group), Z-VAD-fmk (50 μM Z-VAD-fmk), dieckol (100 μM dieckol), or a combination (100 μM dieckol and 50 μM Z-VAD-fmk). Following overnight incubation, 10 μL of WST-1® solution was added to each well, further incubated for 3 h, and then read at 460 nm using an ELISA reader.
Fluorescence microscopy
The cells treated with 80 or 100 μM dieckol were rinsed once with phosphate-buffered saline (PBS) buffer (135 mM sodium chloride, 2.7 mM potassium chloride, 4.3 mM sodium phosphate, 1.4 mM potassium dihydrogen phosphate) and stained by addition of DAPI solution (1 μg/mL) to the plates. After incubation in the dark at 37 °C for 20 min, the cells were rinsed once with methanol. Formation of apoptosomes was observed under an ECLIPSE 50i fluorescence microscope (Nikon, Tokyo, Japan).
FACS analysis
Sub-G1 DNA content was examined using flow cytometry. Briefly, the cells were harvested by trypsinization and fixed with 70 % ethanol overnight at 4 °C. The cells were then resuspended in PBS buffer containing 0.2 mg/mL RNase A and incubated for 1 h at 37 °C. The cells were stained with 40 μg/mL propidium iodide at room temperature for 30 min in the dark. The distribution of sub-G1 DNA was analyzed using a flow cytometer (Becton-Dickinson, Mountain View, CA, USA).
Preparation of whole cell lysate
Hep3B cells were cultured as mentioned above and treated with 80 or 100 μM dieckol for 24 h, and then harvested. The harvested cells were collected by centrifugation and lysed with ice-cold lysis buffer. After incubation on ice for 30 min, the insoluble materials were removed by centrifugation at 14,000 rpm for 20 min. The protein content of the cell lysates was determined by a Protein Quantification Kit (CBB solution®).
Immunoblotting
An aliquot from each sample was boiled with Laemmli buffer for 5 min, and then resolved using 12 % SDS–polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were electrotransferred to a nitrocellulose membrane and blocked in PBST buffer (135 mM sodium chloride, 2.7 mM potassium chloride, 4.3 mM sodium phosphate, 1.4 mM potassium dihydrogen phosphate, and 0.5 % Tween-20) containing 5 % skim milk powder overnight at 4 °C. The blots were probed with the primary antibodies (cytochrome c, Bid, Bak, Bim, AIF, β-actin, cleaved caspase-8, caspase-12, cleaved caspase-3, cleaved caspase-7, cleaved caspase-9, caspase-9, and cleaved PARP). The blots were then washed three times in PBST, followed by incubation for 1 h with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG as the secondary antibody. The blots were then washed in PBST and visualized by ECL detection solutions (Pierce, Rockford, IL, USA).
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) for the indicated number of separate experiments. The mean of the control was compared with the mean of each individual treatment group by one-way ANOVA followed by Tukey’s test using the statistical software Sigma Plot v.12.3 (Systat Software Inc., San Jose, CA, USA), and a statistically significant difference was set at P ≤ 0.001.
Results
Induced death of hepatocellular carcinoma cells by dieckol
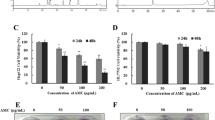
The effect of dieckol on HCC-derived Hep3B, Sk-Hep1 cells and human embryonic kidney HEK293 cells were examined by a cell viability assay using WST-1® solution. The results showed that dieckol did not significantly affect the inhibition of cell proliferation in either HEK293 or Sk-Hep1 cells. In Hep3B cells, however, dieckol treatment inhibited cell viability resulting in induced cell death in a dose-dependent manner. Cell death reached 62.2 % with 100 μM and 18.4 % with 80 μM of dieckol treatment for 24 h, respectively (Fig. 2a), and those concentrations were used in subsequent experiments to investigate the mechanism of cell death. Morphological changes of Hep3B cells treated with or without dieckol were visualized under an inverted microscope (200×) (Fig. 2b).

Effects of dieckol on the proliferation of Hep3B cells. Cell viability was measured by WST-1 cell proliferation assay. Results from 1 of 3 different experiments are shown; data are mean ± SD. Asterisks significant difference from control by ANOVA, *p < 0.001. a Sk-Hep1, Hep3B, and HEK293 cells were incubated with various concentrations (80–140 μM) of dieckol for 24 h. b Hep3B cells were treated with (80 or 100 μM) or without dieckol for 24 h and the images were acquired under inverted microscopy (magnification ×200). c Z-VAD-fmk, a caspase inhibitor, enhances viability of Hep3B cells which are inhibited by dieckol. The cells were treated with the indicated conditions for 24 h followed by WST-1® cell viability assay
The relationship between cells being caspase-dependent and caspase-independent was examined using dieckol with or without Z-VAD-fmk, a caspase inhibitor. Cells were divided into four groups: non-treated, Z-VAD-fmk (treated with 50 μM Z-VAD-fmk), dieckol (treated with 100 μM dieckol), and the combination (treated with both 100 μM dieckol and 50 μM Z-VAD-fmk) (Fig. 2c). In Hep3B cells, the dieckol treatment resulted in the death of 75 % of the cells, and ~45.2 % in the dieckol plus Z-VAD-fmk combination group. The results suggest that dieckol enhances apoptotic cell death in the caspase-dependent pathway.
Detection of apoptotic cells by fluorescence staining
As the death of Hep3B cells induced by dieckol treatment shows characteristics of apoptosis, we detected nuclear condensation and formation of apoptosomes. The cells were stained with DAPI (4′,6-diamidino-2-phenylindole dihydrochloride hydrate) as described and analyzed under an ECLIPSE 50i fluorescence microscope (1,000×). The results of DAPI staining showed totally different patterns in the treated and untreated cells. The cells treated with 80 μM of dieckol showed changes of nuclear membrane such as shrinkage and blebbing. A classical hallmark of apoptotic cells, nuclear condensation (apoptosome formation), was observed in Hep3B cells treated with 100 μM of dieckol for 24 h (Fig. 3a).

Dieckol induced apoptosis in Hep3B cells. The cells were treated with (80 or 100 μM) or without dieckol for 24 h. a DAPI staining: the cells containing condensed chromatin or exhibiting fragmented nuclei are identified as apoptotic bodies. b FACS analysis for quantification of sub-G1 DNA content in the cells. The flow cytometry histograms are representative of three separate experiments
Quantification of sub-G1 DNA content
The quantification of apoptosis was confirmed by estimating the quantity of sub-G1 DNA in dieckol-treated Hep3B cells using flow cytometry analysis. The results indicate that the presence of sub-G1 DNA in the control was ~1.85 %, whereas treated Hep3B cells contained 4.49 % of sub-G1 at 80 μM and 11.6 % at 100 μM of dieckol (Fig. 3b), indicating that the sub-G1 cell population was gradually increased in a dose-dependent manner. As the population of sub-G1 cells indicates the presence of apoptotic cells, the dieckol treatment consequently resulted in an increase in the proportion of apoptotic cells.
The activation of apoptosis-related proteins by dieckol
Immunoblotting showed that dieckol at a dose of 100 μM induces apoptosis in Hep3B cells by releasing cytochrome c from the mitochondria. The expressions of Bid and Bim were up-regulated by the activation of caspase-8, and caused the release of cytochrome c. Dieckol induces expressions of caspase-8, Bid, and Bak in a dose-dependent manner and also induces the dissociation of Bim from LC8 (a component of the microtubule-associated dynein motor complex), and promotes apoptosis (Fig. 4a). The expression of AIF was increased in a dose-dependent manner in dieckol-treated Hep3B cells by cleaved PARP activation. PARP is involved in the DNA repair mechanism that is important for cell viability, and is cleaved by several caspases, particularly cleaved caspase-3 and cleaved caspase-7. In our results, the expression levels of both cleaved caspase-3 and -7 were higher in dieckol-treated cells and caused cleaved PARP to be increased (Fig. 4b).

Expression of apoptotic proteins in dieckol-treated Hep3B cells. The cells were treated with 0, 80 or 100 μM of dieckol for 24 h. Cell lysates were analyzed by Western blotting with the antibodies indicated. a Dieckol modulates the ratio between the amounts of Bid, Bim, cytochrome c, and AIF and b induces activation of the critical molecules such as cleaved caspase-3, 7, 8, 9, 12 and PARP. Photographs show chemiluminescent detection of the blots, which are representative of 3 independent experiments
Discussion
In this study, we demonstrated that dieckol induced the expressions of Bim and Bid through AIF activation and released cytochrome c, leading to apoptosis in Hep3B cells. The extrinsic/death receptor pathway is mediated by cell surface death receptors, such as Fas. The binding of death ligands such as Fas-L to their specific receptors results in the recruitment of the adaptor protein Fas-associated death domain and caspase-8, leading to the cleavage and activation of caspase-8. Activated caspase-8 amplifies the apoptotic signal through either direct activation of downstream executioner caspase-3 or cleavage of Bid (a BH3-only pro-apoptotic Bcl-2 family protein), subsequently leading to the release of cytochrome c [27, 28]. We determined that the expression levels of cleaved caspase-8 and Bid were increased in a dose-dependent manner (Fig. 4). These results indicate that caspase-8 directly promotes caspase-3 activation in dieckol-treated cells, rather than through activating caspase-9 (Fig. 4).
The permeability of mitochondrial membrane is precisely regulated by the Bcl-2 family of proteins [29], which is subdivided into three classes on the basis of their functions and the numbers of Bcl-2 homology (BH) domains present. These classes consist of the anti-apoptotic proteins such as Bcl-2, Bcl-xL, and Mcl-1 which harbor 4 BH domains (BH1–BH4), the pro-apoptotic proteins, including Bax, Bak, and Bok, which harbor 3 BH domains (BH1–BH3), and the BH3-only pro-apoptotic proteins, including Bik, Bim, Bad, Bmf, and Bid, which share homology only within the BH3 domain. These proteins are capable of forming either homo-oligomers or heterodimers with one another and appear to perform distinct functions in the regulation of mitochondrial membrane permeabilization [30]. In the current study, we demonstrated that dieckol increased the expression levels of pro-apoptotic BimS, BimL, and Bid (Fig. 4a). Bim consists of three isoforms; BimS, BimL, and BimEL (Bim-short, Bim-long, and Bim-extra long, respectively) with different intrinsic toxicities by alternative splicing [31], and all play roles in different modes of regulation by various pro-death and pro-survival signaling pathways [32]. Bim also mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis [33]. Our results showed that the increased expression of pro-apoptotic proteins and disruption of the mitochondrial membrane permeability incurred by dieckol promote release of cytochrome c with AIF, and thereby activation of caspase-9 and caspase-3 in Hep3B cells.
In the case of Rat-1 cells, after sufficient release of cytochrome c from the mitochondria into the cytosol, chromatin condensation and nuclear fragmentation (karyorrhexis) by AIF had progressed [34]. Moreover, there is extensive evidence that mitochondria play an essential role in apoptosis by releasing apoptogenic factors, such as cytochrome c and AIF, from the intermembrane space into the cytoplasm, which activates the downstream execution phase of apoptosis [30]. AIF is released from the mitochondria and participates in caspase-independent apoptosis [34]. Apoptosis occurs via the coordinated activation of executioner caspases, which cleave a variety of cellular substrates, including PARP, a nuclear enzyme involved in DNA repair [27]. Therefore, we also investigated the expression of AIF-induced apoptosis-related genes, cleaved PARP, cleaved caspase-3, and cleaved caspase-7. Cell lysates were subjected to immunoblot analysis with antibodies against PARP and the cleaved caspases as indicated, so that the activated form of those genes was increased (Fig. 4b). It has been established that these executioner caspases are activated by initiator caspases, including caspase-8 and -9 [27]. Our results showed that dieckol induces the activation of caspase-8 and -9, resulting in the activation of caspase-7 and -3, which provokes the induction of apoptosis. The activation of caspases is primarily triggered via two distinct pathways: the extrinsic/death receptor pathway and the intrinsic/mitochondrial pathway. The activation of the intrinsic/mitochondrial pathway is involved in the permeabilization of the outer mitochondrial membrane with subsequent releases of pro-apoptotic factors, including cytochrome c, into the cytosol. Cytosolic cytochrome c alters the conformation of the cytosolic protein Apaf-1, whereas this protein oligomerizes with inactive procaspases-9, resulting in the activation of this enzyme [35].
The results presented here indicate, for the first time, that dieckol can increase depolarization of the mitochondrial membrane in Hep3B cells by release of cytochrome c with AIF from the mitochondria, which, in turn, results in activation of caspase-9, subsequently resulting in the activation of caspase-3 and caspase-7. It has been reported that dieckol possesses protective effects on UV-B radiation-induced cell damage in human fibroblasts [36]. As summarized in Fig. 5, our results demonstrated that dieckol induced apoptosis which acts through multiple signaling cascades, through the activation of caspases-3, -7, -8, -9 and PARP on Hep3B cells, a representative p53-null HCC that contains copies of hepatitis B virus (HBV) genomes in their chromosomes and actively secrets HBsAg [37]. Hep3B cells were significantly more sensitive to the growth inhibition and cytotoxicity of dieckol than HEK293 and Sk-Hep1 cells (Fig. 2a). Although there are some risk factors, such as aflatoxin, persistent hepatitis C viral infection, and alcoholic cirrhosis, which cause HCC, HBV, which is a 3.2-kb, partially dsDNA, non-cytopathic virus, is the most important etiologic factor for malignant HCC. This is because patients with a persistent HBV infection in hepatocytes are at a higher risk of developing chronic hepatitis, cirrhosis, and/or HCC [38]. We therefore showed that dieckol from natural sources induces the activation of the extrinsic/death receptor and intrinsic/mitochondrial apoptotic pathways in Hep3B cells.

Proposed dieckol-induced apoptosis signaling pathways in Hep3B cells
References
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108
Ho JW, Man K, Sun CK, Lee TK, Poon RT, Fan ST (2005) Effects of a novel immunomodulating agent, FTY720, on tumor growth and angiogenesis in hepatocellular carcinoma. Mol Cancer Ther 4:1430–1438
Roberts LR (2008) Sorafenib in liver cancer—just the beginning. N Engl J Med 359:420–422
Roayaie S, Blume IN, Thung SN, Guido M, Fiel MI, Hiotis S, Labow DM, Llovet JM, Schwartz ME (2009) A system of classifying microvascular invasion to predict outcome after resection in patients with hepatocellular carcinoma. Gastroenterology 137:850–855
Newell P, Villanueva A, Llovet JM (2008) Molecular targeted therapies in hepatocellular carcinoma: from pre-clinical models to clinical trials. J Hepatol 49:1–5
Pang RW, Poon RT (2007) From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology 72(Suppl 1):30–44
Varghese L, Agarwal C, Tyagi A, Singh RP, Agarwal R (2005) Silibinin efficacy against human hepatocellular carcinoma. Clin Cancer Res 11:8441–8448
Kim AR, Shin TS, Lee MS, Park JY, Park KE, Yoon NY, Kim JS, Choi JS, Jang BC, Byun DS, Park NK, Kim HR (2009) Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and anti-inflammatory properties. J Agric Food Chem 57:3483–3489
Ragan MA, Glombitza KW (1986) Phlorotannins, brown algal polyphenols. Prog Phycol Res 4:129–241
Zou Y, Qian ZJ, Li Y, Kim MM, Lee SH, Kim SK (2008) Antioxidant effects of phlorotannins isolated from Ishige okamurae in free radical mediated oxidative systems. J Agric Food Chem 56:7001–7009
Kang KA, Lee KH, Chae S, Koh YS, Yoo BS, Kim JH, Ham YM, Baik JS, Lee NH, Hyun JW (2005) Triphlorethol-A from Ecklonia cava protects V79-4 lung fibroblast against hydrogen peroxide induced cell damage. Free Radic Res 39:883–892
Kang KA, Lee KH, Park JW, Lee NH, Na HK, Surh YJ, You HJ, Chung MH, Hyun JW (2007) Triphlorethol-A induces heme oxygenase-1 via activation of ERK and NF-E2 related factor 2 transcription factor. FEBS Lett 581:2000–2008
Okada Y, Ishimaru A, Suzuki R, Okuyama T (2004) A new phloroglucinol derivative from the brown alga Eisenia bicyclis: potential for the effective treatment of diabetic complications. J Nat Prod 67:103–105
Yoon NY, Chung HY, Kim HR, Choi JS (2008) Acetyl- and butyrylcholinesterase inhibitory activities of sterols and phlorotannins from Ecklonia stolonifera. Fish Sci 74:200–207
Kang K, Park Y, Hwang HJ, Kim SH, Lee JG, Shin HC (2003) Antioxidative properties of brown algae polyphenolics and their perspectives as chemopreventive agents against vascular risk factors. Arch Pharm Res 26:286–293
Fukuyama Y, Kodama M, Miura I, Kinzyo Z, Mori H, Nakayama Y, Takahashi M (1990) Anti-plasmin inhibitor. Structure of phlorofucofuroeckol A, a novel phlorotannin with both dibenzo-1,4-dioxin and dibenzofuran elements, from Ecklonia kurome Okamura. Chem Pharm Bull 38:133–135
Ryu BM, Li Y, Qian ZJ, Kim MM, Kim SK (2009) Differentiation of human osteosarcoma cells by isolated phlorotannins is subtly linked to COX-2, iNOS, MMPs, and MAPK signaling: implication for chronic articular disease. Chem Biol Interact 179:192–201
Joe MJ, Kim SN, Choi HY, Shin WS, Park GM, Kang DW, Kim YK (2006) The inhibitory effects of eckol and dieckol from Ecklonia stolonifera on the expression of matrix metalloproteinase-1 in human dermal fibroblasts. Chem Pharm Bull 29(8):1735–1739
Glombitza KW, Vogels HP (1985) Antibiotics from algae. XXXV Phlorotannins from Ecklonia maxima. Planta Med 51:308–312
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376:37–43
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347
Salvesen GS, Dixit VM (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase. Cell 81:801–809
Tu H, Jacobs SC, Borkowski A, Kyprianou N (1996) Incidence of apoptosis and cell proliferation in prostate cancer: relationship with TGF-beta 1 and Bcl-2 expression. Int J Cancer 69:357–363
Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS (2004) Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res 64:8804–8807
Tian Z, Pan R, Chang Q, Si J, Xiao P, Wu E (2007) Cimicifuga foetida extract inhibits proliferation of hepatocellular cells via cell cycle arrest and apoptosis. J Ethnopharmacol 114:227–233
Jin Z, El-Deiry WS (2005) Overview of cell death signaling pathways. Cancer Biol Ther 4:139–163
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281:1305–1308
Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2:647–656
Breckenridge DG, Xue D (2004) Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol 16:647–652
Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM (2001) Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm Genome 12(2):163–168
Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, Wong KK, Letai A (2007) Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res 67:11867–11875
Rasheva VI, Domingos PM (2009) Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 14:996–1007
Candé C, Cecconi F, Dessen P, Kroemer G (2002) Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci 115:4727–4734
Antonsson B, Martinou JC (2000) The Bcl-2 protein family. Exp Cell Res 256:50–57
Heo SJ, Ko SC, Cha SH, Kang DH, Park HS, Choi YU, Kim DK, Jung WK, Jeon YJ (2009) Effect of phlorotannins isolated from Ecklonia cava on melanogenesis and their protective effect against photo-oxidative stress induced by UV-B radiation. Toxicol In Vitro 23:1123–1130
Twist EM, Clark HF, Aden DP, Knowles BB, Plotkin SA (1981) Integration pattern of hepatitis B virus DNA sequences in human hepatoma cell lines. J Virol 37(1):239–243
Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV (1998) Immune pathogenesis of hepatocellular carcinoma. J Exp Med 188(2):341–350
Acknowledgments
This work was financially supported by the Ministry for Food, Agriculture, Forestry and Fisheries, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yoon, JS., Kasin Yadunandam, A., Kim, SJ. et al. Dieckol, isolated from Ecklonia stolonifera, induces apoptosis in human hepatocellular carcinoma Hep3B cells. J Nat Med 67, 519–527 (2013). https://doi.org/10.1007/s11418-012-0709-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-012-0709-0