
Abstract
Purpose
Arbuscular mycorrhizal (AM) fungi represent a functionally important component of soil microbial communities. It is critical to achieve an improved understanding of the community structure of the indigenous AM fungi if we are to use this group of fungi either as indicators of ecosystem health or to enhance the sustainability of agricultural systems. In the present study, we assessed the impact of land use and soil depth on the number and community composition of infective AM fungal propagules in high-input agricultural ecosystems.
Materials and methods
Three different agricultural land use systems with high management intensity were selected, i.e., vegetable greenhouses, arable land, and open vegetable fields. Soil samples were collected from different soil depths (0–30, 30–60, and 60–90 cm) of three replicated (triplicate) land-use types at two sites in north China. The modified mean infection percentage (MIP) method was used to determine the inoculum potential of each soil sample. The community composition of AM fungi was analyzed using PCR, cloning, and sequencing techniques.
Results and discussion
Land use and soil depth greatly influenced the development of root-associated AM fungi and also edaphic properties. MIPs and species richness were lower in the top (0–30 cm) of soil profile in the vegetable greenhouses than in the nearby open vegetable fields or arable fields. Moreover, AM fungal distribution in the soil profile varied with land use. The MIPs and species richness decreased with increasing soil depth in the arable land and open vegetable fields. By contrast, in the greenhouse vegetable soils, the highest richness was found in the deepest soil depth (60–90 cm). The presence and prevalence of AM fungi also varied with land-use type and soil depth. Of the 13 total phylotypes investigated, Glo 6, Div 1, and Glo 1 accounted for 82.8 % of AM fungal clones analyzed and were present in each land-use type and soil depth. There were some observed phylotypes that were only found below 30 cm depth (Glo 4, Glo 5, Div 3, and Div 4).
Conclusions
AM fungal infectivity and richness were not always lower in the deeper soil profiles in the greenhouse than in the agricultural fields. High land-use intensity was correlated with a preferential persist of some AM fungi in the deeper soil layers to escape from the adverse conditions caused by intensive farming practices.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Arbuscular mycorrhizal (AM) fungi are a main component of the soil microbiota in most agroecosystems and they form symbiotic associations with a majority of plant species including nearly all crop species (Newman and Reddell 1987; Simon et al. 1993; Jansa et al. 2006). AM fungi are known to increase plant growth by facilitating nutrient uptake from the soil via an extensive extraradical mycelium (Li et al. 1991a,b) and increase the tolerance of plants to a range of biotic and abiotic stresses (Meharg and Cairney 2000; Augé 2001; Borowicz 2001; Pozo and Azcón-Aguilar 2007). It is evident from the beneficial effects that AMF are crucial for the functioning of terrestrial ecosystems (Smith and Read 2008).
However, modern intensive farming practices such as tillage, chemical pesticide, and fertilizer use may reduce the abundance and diversity of AM fungi (Abbott and Robson 1991; Helgason et al. 1998; Oehl et al. 2003, 2004). Such management practices can result in a certain reduced subset of AM fungal taxa and other soil organisms (Johnson et al. 1992; 1993; Kiers et al. 2002). Nevertheless, some studies still suggested this decrease can be avoided to maintain the diversity of AM fungi in arable soils as a possible agricultural resource of the future (Franke-Snyder et al. 2001; Hijri et al. 2006; García et al. 2007; Lekberg et al. 2008; Martinez and Johnson 2010).
Although there have been numerous studies on AM fungal diversity in intensively managed agricultural ecosystems (Oehl et al. 2004, 2005, 2010; Köhl et al. 2014), only a few studies have described AM fungal diversity in intensively managed systems such as greenhouse vegetable production systems (Jiao et al. 2011). In China, the total area devoted to vegetable production has increased to over 18.2 million ha over the last two decades and accounted for 11.6 % of the total agricultural area in 2007 (China Ministry of Agriculture 2008). The high market value of vegetables has encouraged farmers to use large amounts of organic manures and mineral fertilizers. Nutrient application rates are considerably higher in vegetable production systems than in arable systems. For example, in Shouguang County, Shandong Province, more than 65 % of the arable land is used for intensive greenhouse production. This area is supplied with 2220 kg fertilizer N ha−1 and 1,800 mm irrigation water per year (Zhu et al. 2005; He et al. 2007). Furthermore, in north China, specific vegetables are grown in continuous cropping in some areas. Whether AM fungi can persist in such high-input and intensively cultivated systems and re-inoculate their host plants successfully is not well characterized.
Previous studies on AM fungal abundance and AM fungal community structure are generally restricted to the main rooting zone (10–30 cm soil depth; Redecker 2002; Vandenkoornhuyse et al. 2002; Jansa et al. 2003; Johnson et al. 2003). However, work by Oehl et al. (2005) indicates that an AM fungi “gene bank” might persist in the subsoil, which may facilitate agro-ecological restoration when a high-input farming system is switched to a low-input system since it must rely more on internal biotic and abiotic resources. Therefore, in the present study, we sought to test the infectivity and composition of the indigenous AM fungi to understand how they are distributed in the soil profile in high-input agricultural ecosystems.
In general, studies characterizing AM fungal communities in field soils are based on the analysis of spores, which can sometimes be complemented by trap culturing (Douds et al. 1993; Oehl et al. 2004, 2010). Spore-based surveys are considered to be the baseline for assessing the impact of agricultural practices on AM fungal communities (Douds and Millner 1999). However, morphological characterization of AM fungal spores and their diversity might not reflect the actual functional symbiosis that facilitates the formation of active fungal structures within and outside roots (Clapp et al. 1995; Jansa et al. 2003). These active structures, such as fungal hyphae in the roots and soil as well as arbuscules within roots, can only be identified accurately using molecular or immunological approaches (Redecker et al. 2003; Sanders 2004). The present study investigated the molecular diversity of AM fungi at three soil depths from the surface soil to the subsoil in areas with three types of high-input land use.
2 Materials and methods
2.1 Sites and agricultural practices
The study sites, namely, Taiyuan (T) and Shouguang (S), are located at the middle and lower reaches, respectively, of the Yellow River Basin, one of the most important agricultural zones in China, which accounts for 12.4 % of the cultivated land area and 7.7 % of total grain production. Although these percentages are low the area is an important ecological barrier in north China. T and S have yearly precipitation of 456 and 594 mm, annual average temperatures of 9.5 and 12.7 °C, and yearly sunshine of 2808 and 2549 h, respectively. At site T, three typical intensive cropping systems were studied, i.e., vegetable greenhouses, open vegetable fields, and arable land. At site S, only vegetable greenhouses and arable land were studied because typical open vegetable land was absent from this area. The three selected land-use types are common at the target area and the land management practices at these plots reflect current practices used in the region. The plot details are shown in Table 1. Rooting depths of crop roots can extend to 90 cm depth, while tilling only penetrates the soil to a depth of 30 cm. Then, the current soil sampling depths extended to 90 cm and samples were sectioned into 0–30, 30–60, and 60–90 cm depth categories according to the local tillage regimes and plant root distribution range.
2.2 Soil sampling, preparation, and chemical analysis
Soil samples collected from arable land and open vegetable fields system at sites T and S were obtained from March to April 2010 and in the vegetable greenhouse systems at harvest time of the winter–spring season (May–June 2010). Three replicate plots of open vegetable fields and arable lands were selected in villages that had all three (or two) land uses for sampling (plot size 102–120 m2). Three typical commercial greenhouses at each site (100 m × 8 m at site T and 84 × 8.5 m at site S), constructed of clay walls and covered with polyethylene film, were randomly selected in each selected village.
We used a stratified random sampling approach to collect individual soil cores. In each of the three replicate plots (or greenhouse) per field site, six soil cores were drilled to a depth of 90 cm using an 8-cm-diameter soil corer and the cores were separated in sections corresponding to 0–30, 30–60, and 60–90 cm soil depth. Soils were sieved on-site through a 4-mm mesh, and then all six of the cores from a given plot (or greenhouse) that corresponded to a certain soil depth were homogenized by hand into a single composite sample per plot (or greenhouse). A subsample of 200 g soil was carefully ground by hand from each of the 45 pooled composite samples, thoroughly mixed, and air-dried. They were stored at 4 °C until analysis for pH, organic carbon, and available P. Additional subsamples of 500 g soil were stored in self-sealing plastic bags at 4 °C and used for the bioassay of inoculum potential.
Soil physical and chemical properties were measured according to standard methods, i.e., pH (1:2.5 soil/water), soil total N (Kjeldahl method), soil total P (H2SO4–HClO4 digestion; Mo–Sb colorimetric method), available P (Olsen and Sommers 1982), and organic matter content (Cambardella et al. 2001).
2.3 Bioassay for inoculum potential
The bioassay for inoculum potential was performed using a modified mean infection percentage (MIP) method (Richter et al. 2002). Soil samples were sieved to 2 mm (large root fragments were cut to 1 cm and returned to the soil) and diluted in a 1:3 ratio (v/v) with autoclaved no. 20 silica sand (121 °C for 1 h). Each soil dilution was used to fill six containers (3.8 × 14 cm, 104 ml), giving a total of 270 containers. Cucumber (Cucumis sativus L.) seeds were surface sterilized in 10 % bleach for 3 min, and two seeds were sown in each container and covered with a 1-cm layer of autoclaved no. 12 sand. The containers were placed in a greenhouse in a randomized design under natural light conditions and day/night temperatures of 31/22 °C. After emergence, the seedlings were thinned to one per container.
The plants were harvested at 30 days. Roots were washed free of soil and cut into 1-cm segments. One half of the roots were stored at −20 °C for molecular analysis, and the remainders were stored at 4 °C for MIP determination. Approximately 0.5 g fresh roots per sample was stained with Trypan blue. Root segments (100 of each replicate) were mounted on glass microscope slides and examined under a light microscope for infection. A segment was considered infected if it contained hyphae plus arbuscules, coils, or vesicles. Percentage infection was recorded as the number of infected segments per 100 root segments (Moorman and Reeves 1979).
2.4 DNA extraction from roots and nested-PCR for AM fungi
The present study focused on the AMF with vital AMF propagules in the field. AM fungal community was therefore analyzed using root samples from the MIP bioassay. A subsample (~0.5 g) of the stored roots was cut into 0.5-cm pieces and immediately frozen in liquid nitrogen. Genomic DNA was extracted by a modification of the CTAB method (Li 2000; Li et al. 2014). Volumes of 2–5 μL of each DNA extract were used as polymerase chain reaction (PCR) templates.
A two-step procedure (nested PCR) was conducted using the primer pairs SSUmAf–LSUmAr and SSUmCf–LSUmBr (Krüger et al. 2009). The target region for PCR included the partial sequences of LSU rDNA and SSU rDNA and the whole ITS rDNA. The first amplification was performed using Ar/Af. The 25-μL reaction volume contained 2 μL 10× PCR buffer, 0.2 mM dNTPs, 0.5 mM of each primer, 1 U Taq polymerase (Trans-easy taq), and 1 μL DNA template. The PCR program was as follows: initial denaturation at 94 °C for 5 min, 30 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 1 min, and extension at 72 °C for 1.5 min, followed by extension at 72 °C for 10 min. The quantity of PCR products was estimated by conducting 1 % agarose gel electrophoresis using 5 μL PCR products. Next, 1 μL of PCR products was used directly as a template for the second PCR that was conducted using Cf/Br under the reaction conditions described above. Nested PCR products were separated by 1 % agarose gel electrophoresis and purified using an Extraction Kit (BioTeke, Beijing, China) according to the manufacturer’s instructions.
2.5 Cloning, sequencing, and construction of clone libraries
The PCR products from roots of the replicated six containers were pooled (totally 45 samples) and cloned into pGEM-T Easy Vector System (Promega) and transformed into competent cells of Escherichia coli DH-5α strain following the manufacturer’s protocol. Three replicates of each land use were grouped into one clone library, and 15 clone libraries for AM fungi were constructed from all soil depths of each land use. Clones containing the converted DNA fragments were selected by blue/white screening, and eventually, 80 white colonies were used and grown overnight at 37 °C in liquid Luria–Bertani medium (5 g yeast extract, 10 g tryptone, 10 g NaCl, and deionized water added to a final volume of 1 L, pH 7.0) containing ampicillin (50 μg mL−1). Thereafter, the clones were identified by PCR with primer pair T7/SP6.
2.6 Restriction analysis, sequencing, and taxonomic classification
To roughly detect intrasporal and intersporal sequence variability in the clones, PCR products of 64 positive clones in each library were digested with the restriction enzymes Hinf and MboІ (Fermentas, USA; Krüger et al. 2009) for 5 h to produce restriction fragment length polymorphism profiles according to the manufacturer’s protocols. Restriction fragments were separated at 100 V for 1.5 h in 1× TAE buffer in 2 % (w/v) agarose gel, stained with ethidium bromide (0.5 μg ml−1), visualized under 300 nm UV light, and photographed. Representatives of each RFLP type from each sample were sequenced (Liu et al. 2001) by Beijing ZhongKeXiLin Biotechnology Company (Beijing, China).
The sequence data obtained were checked with the BLAST tool at GenBank to determine whether sequences were derived from Glomeromycota (based on the origin of the best scoring hit in the GenBank).
2.7 Sequence analysis
DNA sequences were edited with the SeqMan program of the Lasergene Package (DNAStar Inc., Madison, WI, USA). Before processing the sequences for phylogenetic analysis, representative sequences were targeted to define the divergent sequences from the same species using the DOTUR program (Schloss and Handelsman 2005). A dataset containing these representative sequences and reference sequences obtained from GenBank broadly representing the Glomeromycota was constructed. These sequences were aligned using Megalign program of the Lasergene Package, and the alignment was used to generate tree topologies. Phylogenetic trees were constructed by distance analysis using the neighbor-joining (NJ) algorithm in Paup 4.0 using the Kimura two-parameter model. Confidence in specific clades from the resulting topology was tested by bootstrap analysis with 1000 replicates. Branches corresponding to partitions reproduced in <70 % bootstrap replicates were collapsed. Sequence phylotypes were therefore defined in a conservative manner as consistently separated monophyletic groups in the phylogenetic tree. All the gene sequences in this phylogenetic tree have been deposited in GenBank under accession numbers JX683733 to JX683763.
2.8 Statistical data analysis
The sampling effort in each library was evaluated by calculating the Good coverage (C) according to the equation C = 1 − (n/N), where n is the number of representative sequences and N is the total number of clones analyzed in the corresponding library (Good 1953). AM fungal richness was assessed as the number of different phylotypes detected in a sample.
Significance of differences in soil properties, species richness, and MIP between the samples from different soil depths and land uses at a given site was tested using Fisher’s least significant difference (LSD) at P < 0.05 after a one-way analysis of variance (ANOVA). The relationships between AM fungi and soil chemical parameters were examined with Pearson’s correlation coefficients. Dependency of MIP and fungal richness on selected soil parameters was confirmed using stepwise linear regression analysis. The SPSS 13.0 software package was used to conduct all statistical analysis.
To explore the relationship between environmental variables and AMF community composition, the environmental variables (including soil depths and soil properties) were fitted as vectors onto the nonmetric multidimensional scaling (NMDS) plot using function “envfit” in the R version (R Development Core Team 2011). To test whether different soil types from sites T and S harbor different community composition, soil types were fitted as ellipse onto NMDS plot using function “ordiellipse” from “vegan” library in the R.
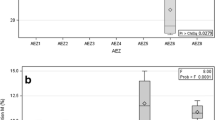
The present study assessed the AM fungal community within the roots of the MIP assay and not directly from the roots or bulk soil collected from the field. The MIP assay is often used to assess the inoculum potential of AM fungi in the field. Thus, some highly mycorrhiza-dependent plant species are used as the host plant, such as maize and clover. When Richter et al. (2002) assessed MIP from abandoned agricultural fields and semi-arid grasslands in riparian floodplains, they selected corn (Zea mays L.) as the host plant. Cucumber is a highly mycorrhiza-dependent vegetable (Jakobsen and Rosendahl 1990), and we therefore selected it as the host plant in the present study. We also tried to extract DNA directly from the roots and bulk soil in a vegetable greenhouse at site S. Most of samples from 60 to 90 cm soil depth failed (not published data). The low MIP values also provide convincing evidence that there were very few propagules there (Fig. 1). Compared to the bias from cultures, we consider that the bias involved in extracting DNA directly from a small sample of roots and bulk soil is very high. This method might be helpful in the evaluation of the AM fungal community under adverse conditions.

Mycorrhizal inoculum potential (MIP) under different land-use types and soil depth categories at the three sites studied. Values are mean ± 1 SE. Lowercase letters represent differences within soil depth. Uppercase letters represent differences among land-use types. All differences are at the significance level P < 0.05
3 Results
3.1 Edaphic properties
Soil properties were found to vary with land use and soil depth (Table 2, Table S2). Vegetable greenhouse soils (0–30 cm) had approximately three times the total P of arable fields at site S and about twice of arable land at site T. Likewise, Olsen P in vegetable greenhouses (0–30 cm) was more than ten times greater than in arable soil at site S and eight times greater than at site T. Both total and Olsen P in open vegetable fields was at an intermediate level for all the land-use types (Table 2). In most cases, nutrients (organic matter, total N, P, and Olsen P) in the topsoil (0–30 cm) were considerably higher than in the intermediate and/or the deepest layer but pH was often lowest in the topsoil (Table 2).
3.2 MIP
Bioassay results indicate that the number of infective propagules differed according to the land-use type and soil depth (Fig. 1, Table S2). The highest MIP value was noted in the top soil obtained from arable fields at both sites (Fig. 1a, b). In the vegetable greenhouses, there was no significant difference of propagules among soil depths at both sites. In contrast, the topsoil contained considerably more propagules and showed a decreasing trend with increasing soil depth under the other two land uses (Fig. 1a, b).
3.3 Molecular analysis of AM fungal sequences
Target rDNA sequences from all root samples of the MIP assay were successfully amplified by nested PCR. A total of 2880 clones from 15 libraries were screened by RFLP and 492 clones were subsequently sequenced (two clones of each RFLP type). Preliminary BLAST searches revealed 481 sequences (2767 clones) belonging to AM fungi, excluding non-AM fungi (mostly were uncultured Zygomycetes, uncultured Ascomycete clones and Zygomycete spp.) and chimeric sequences (113 clones). These non-AM fungi and chimeric sequences were excluded from sequence analyses. Each library provided good coverage above 97.5 %, with an integrated value of 98.9 %, which suggests that the majority of fungal species in the 15 libraries were selected. Partial LSU rDNA sequences were grouped into 31 different OTUs on the basis of sequence similarities (95–100 %) using the DOTUR program (Table S1).
The topology of the phylogenetic tree and the results of BLAST indicate that all the AM fungal sequences were clustered into 13 discrete groups or phylotypes with an NJ bootstrap support of ≥75 %. The 13 sequence phylotypes were grouped into three families: Glomeraceae, Diversisporaceae, and Paraglomeraceae (Fig. 2). Most sequences appeared to represent Glomus, Funneliformis, Rhizophagus, Paraglomus, and Diversispora. Five clusters (Glo 4, 5, 7, and Div 3, 4) were likely not related to any sequences of AM fungi in culture or previously reported in the database. Clone numbers indicate the overall dominance of Glo 6 (31.4 % of all clones isolated), Div 1 (30.5 % of all clones isolated), and Glo 1 (20.9 % of all clones isolated) sequence groups. No other sequence phylotypes contributed to more than 10 % of clones (Fig. 2).

Neighbor-joining phylogram of AMF sequences obtained from plant roots in the MIP bioassay. Number above the nodes indicates the bootstrap support in neighbor-joining analysis. Asterisk Relative abundance of the particular phylotype in the investigation
3.4 AM fungal composition and diversity
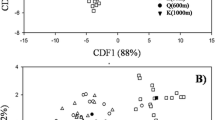
AM fungal community composition varied with land use and soil depth. NMDS ordination yielded sample groupings according to land uses at specific sites (Fig. 3). Of the 13 total phylotypes, the most common types (Glo 6, Div 1, and Glo 1) accounted for 82.8 % of AM fungal clones analyzed and were present in all the soil depths and land-use types examined (Fig. 2, Table 3).

Joint plot of NMDS ordination of AMF communities in soils from sites T and S and the vectors of significant (P < 0.05, except elevation, p = 0.58) environmental variables across sites. The stress value reflects how well the ordination summarizes the observed distances among samples. The length of the arrow is proportional to the strength of correlation between environmental variable and community composition dissimilarities. Blue circles Samples from arable field at S (SA), red triangles samples from greenhouse vegetable soil at S (SG), yellow squares samples from arable land at T (TA), blue stars samples from greenhouse vegetable soil at T (TG), green diagonal squares samples from open vegetable field at T
Almost 40 % (5 out of 13) phylotypes were unique to a specific land-use type or soil depth. For example, Glo 4 was found only in the deepest layer of greenhouse vegetable soils at both sites, and Div 3 was found only in the middle soil depth of arable land. Glo 4, Glo 5, Div 3, and Div 4 were found only below the plough layer (below 30 cm; Table 3).
Richness of AM fungal phylotypes varied with land-use type and soil depth (Fig. 4, Table S2). At both sites, richness was much higher in the 0–30 and 30–60 soil depth of arable land than in the greenhouse vegetable soils. AM fungi were most numerous in the deepest layer of the greenhouse vegetable soils, while the topsoil contained considerably more AM fungi in the other two land-use types (Fig. 4a, b).

Richness detected for each AMF phylotype at different soil depths and land-use categories. Values are mean ± 1 SE. Lowercase letters represent differences within soil depth. Uppercase letters represent differences among land-use types. All differences are at the significance level P < 0.05
3.5 Effects of soil properties on MIP, species richness, and composition
Correlation analysis shows that all soil property indices were strongly related to each other. Using multiple stepwise regression analysis, the infectivity of AM fungi was strongly correlated with the changes in soil Olsen P (negative effect) and total N content (positive effect) across the landscape investigated (R 2 = 0.387, p < 0.001). Only soil pH was negatively correlated with richness (R 2 = 0.371, p = 0.016).
Seven environmental factors fitted as vectors onto the NMDS plot showed that three factors, i.e., land-use type (R 2 = 0.426, p < 0.001), soil depth (R 2 = 0.334, p < 0.001), and soil pH (R 2 = 0.270, p < 0.001) were significantly correlated with AM fungal community composition (Fig. 3).
4 Discussion
We know that land-use change can have significant and long-lasting effects on soil carbon and nutrient contents, soil texture, and pH (Post and Mann 1990; Murty et al. 2002) that arise largely from changes in plant species composition and management practices. Here, we have observed significant differences in nutrient concentrations (total P and Olsen P) between land-use types and soil depths. However, a lack of consistent land-use effects on edaphic factors such as soil pH, soil organic matter, and total N content was also found. Lauber et al. (2008) also demonstrated that some soil properties (soil texture and nutrient status) differed significantly across land-use types, but other edaphic factors (e.g., pH) did not vary consistently with land use. The higher N and P contents and low pH of the greenhouse vegetable soils may be attributable to very high inputs of fertilizers and manures incorporated into the soil to maintain high vegetable yields (Qiu et al. 2010). With the sole exception of pH at site S, soil depth consistently influenced soil edaphic factors (soil pH, C, N, and P) at the two sites, which is in agreement with the results of Ju et al. (2007).
Land use and soil depth also greatly influence the development of root-associated AM fungi. The MIPs in topsoils of open, greenhouse vegetable and arable lands ranged from 30 to 70 %, which was similar to the top 20 cm layer of paired grassland and abandoned agricultural fields in southeastern Arizona (Richter et al. 2002). The MIPs in greenhouse vegetable soils varied greatly among the sites (Fig. 1), ranging from 0 to 21 %, and the explanation for this is not clear. The different climatic conditions, soil types, and crop rotation history of the sites would have been expected to make some impact on the variance of MIPs. Nevertheless, the results demonstrate the persistence of a certain number of AM fungi in the highly intensified land-use types.
High inputs of fertilizer have often been found to be correlated with a decrease in AM fungal species richness and infectivity (Douds et al. 1993; Douds and Johnson 2004; Kurle and Pfleger 1994; Oehl et al. 2004; Verbruggen et al. 2010). Results of the present investigation also show that AM fungal propagule abundance and phylotype richness were lowest in the greenhouse vegetable soils (Figs. 1 and 4). One of the possible explanations for this decline is that greenhouse soils experience a lack of diversity of host plant species. The application of fungicides and pesticides, together with the application of large quantities of fertilizers and especially phosphorus also can inhibit the growth, development, and functioning of AM fungi (Tawaraya et al. 1994; Oehl et al. 2003, 2004, 2005). Stepwise regression analysis showed that AM fungal infectivity was strongly negatively correlated with soil Olsen P content across the landscapes investigated. Some other factors such as low temperature and low light levels inside the vegetable greenhouses are also disadvantageous to AM fungal development (Smith and Read 2008).
Some studies have shown that niche partitioning can maintain AM fungal communities in intensively managed agricultural ecosystems (Jakobsen and Nielsen 1983; Kabir et al. 1998; Neville et al. 2002). We investigated AM fungi in the soil over a large depth range (0–90 cm) and found the result: The presence and prevalence of AM fungi varied with soil depth. This was particularly striking in the greenhouse vegetable soils where AM fungal richness was much higher at the deepest soil depth studied than the top. The distribution of AM fungi may have been driven by comprehensive forces such as soil physical and chemical properties and pressures of oxygen and root distribution (Anderson et al. 1987; Brady and Weil 1996; Entry et al. 2002), which were favorable to AM fungi living in surface soil or deeper in the profile.
Management practices associated with conventional agriculture might favor few AM fungal taxa tolerating the adverse conditions (Entry et al. 2002; Hijri et al. 2006; Chagnon et al. 2013). Studies have shown that frequently tilled agricultural soils tend to be dominated by species belonging to the Glomeraceae (e.g., Helgason et al. 1998; Jansa et al. 2002; Maherali and Klironomos 2012). These species are sometimes called “typical AM fungi of arable lands” or AM fungal “generalists” (Oehl et al. 2003) or even AM fungal “weed” species. In the present study, Glomeraceae accounted for 61.9 % of AM fungal clones in the soils investigated. Phylogenetic analysis suggests that Glomus mosseae (Glo 6), which is related to Glomus caledonium, was the most frequently isolated species in our study, followed by a Diverspora phylotype (Div 1) and Glomus intraradices (Glo 1). Daniell et al. (2001) also found that G. mosseae was the most frequently found phylotype in the roots of arable crops.
Some AM fungal species were only found in a specific land-use type or soil depth. For example, at both sites, Glo 4 was found only at 60–90 cm depth in greenhouse vegetable soils, and Div 3 was found only in the middle soil depth (30–60 cm) of arable land. The possibility of the survival of sensitive AM fungal species in the subsoil has important implications. They might facilitate agro-ecological restoration when switching from a high- to a low-input farming system.
5 Conclusions
The results presented here indicate that AM fungal diversity and community composition strongly depended on land use and soil depth. The vertical distribution pattern of AM fungal infectivity and richness in greenhouse vegetable soils were quite different from agricultural soils. These findings may be useful in understanding how to control intensively managed agricultural ecosystems to make them both sustainable and productive.
References
Abbott LK, Robson AD (1991) Factors influencing the occurrence of vesicular–arbuscular mycorrhizas. Agric Ecosyst Environ 35:121–150
Anderson EL, Millner PD, Kunishi HM (1987) Maize root length density and mycorrhizal infection as influenced by tillage and soil phosphorus. J Plant Nutr 10:1349–1356
Augé RM (2001) Water relations, drought and vesicular–arbuscular mycorrhizal symbiosis. Mycorrhiza 11:3–42
Borowicz VA (2001) Do arbuscular mycorrhizal fungi alter plant–pathogen relations? Ecology 82:3057–3068
Brady NC, Weil RR (1996) Organisms and ecology of the soil. The nature and properties of soil, 11th edn. Prentice-Hall, Upper Saddle River, pp 328–360
Cambardella CA, Gajda AM, Doran JW, Wienhold BJ, Kettler TA (2001) Estimation of particulate and total organic matter by weight loss on ignition. In: Lal R, Kimble JM, Follett RF, Stewart BA (eds) Assessment methods for soil carbon. Lewis, Boca Raton
Chagnon PL, Bradley RL, Maherali H, and Klironomos JN (2013) A trait-based framework to understand life history of mycorrhizal fungi. Trends Plant Sci 18:484–491
China Ministry of Agriculture, 2008. China Agriculture Statistical Data. China Agriculture Press, Beijing, China,p. 16 (in Chinese)
Clapp JP, Young JPW, Merryweather J, Fitter AH (1995) Diversity of fungal symbionts in arbuscular mycorrhizas from a natural communitiy. New Phytol 130:259–265
Daniell TJ, Husband R, Fitter AH, Young JPW (2001) Molecular diversity of arbuscular mycorrhizal fungi colonising arable crops. FEMS Microbiol Ecol 36:203–209
Development Core Team R (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Douds DD, Johnson NC (2004) Contributions of arbuscular mycorrhizas to soil biological fertility. In: Abbott LK, Murphy DV (eds) Soilbiological fertility: a key to sustainable land use in agriculture. Springer, Berlin, pp 129–162
Douds DD, Millner P (1999) Biodiversity of arbuscular mycorrhizal fungi in agroecosystems. Agric Ecosyst Environ 74:77–93
Douds DD, Janke RR, Peters SE (1993) VAM fungus spore populations and colonization of roots of maize and soybean under conventional and low-input sustainable agriculture. Agric Ecosyst Environ 43:325–335
Entry JA, Rygiewicz PT, Watrud LS, Donnelly PK (2002) Influence of adverse soil conditions on the formation and function of arbuscular mycorrhizas. Adv Environ Res 7:123–138
Franke-Snyder M, Douds DD, Galvez L, Phillips JG, Wagoner P, Drinkwater L, Morton JB (2001) Diversity of communities of arbuscular mycorrhizal (AM) fungi present in conventional versus low-input agricultural sites in eastern Pennsylvania, USA. Appl Soil Ecol 16:35–48
García JP, Wortmann CS, Mamo M, Drijber RA, Quincke JA, Tarkalson D (2007) One-time tillage of no-till: effects on nutrients, mycorrhizae, and phosphorus uptake. J Agron 99:1093–1103
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
He FF, Chen Q, Jiang RF, Chen XP, Zhang FS (2007) Yield and nitrogen balance of greenhouse (Lycopersicum esculentum Mill) with conventional and site-specific nitrogen management in Northern China. Nutr Cycl Agroecosyst 77:1–14
Helgason T, Daniell T, Husband R, Fitter AH, Young J (1998) Ploughing up the wood-wide web? Nature 394:431
Hijri I, Sykorová Z, Oehl F, Ineichen K, Mäder P, Wiemken A, Redecker D (2006) Communities of arbuscular mycorrhizal fungi in arable soils are not necessarily low in diversity. Mol Ecol 15:2277–2289
Jakobsen I, Nielsen NE (1983) Vesicular-arbuscular mycorrhiza in field-grown crops. I. Mycorrhizal infection in cereals and peas at various times and soil depths. New Phytol 93:401–413
Jakobsen I, Rosendahl L (1990) Carbon flow into soil and external hyphae from roots of mycorrhizal cucumber plants. New Phytol 115:77–83
Jansa J et al (2002) Diversity and structure of AMF communities as affected by tillage in a temperate soil. Mycorrhiza 12:225–234
Jansa J, Mozafar A, Kuhn G, Anken T, Ruh R, Sanders IR, Frossard E (2003) Soil tillage affects the community structures of mycorrhizal fungi in maize roots. Ecol Appl 13:1164–1176
Jansa J, Wiemken A, Frossard E (2006) The effects of agricultural practices on arbuscular mycorrhizal fungi. In: Frossard E, Blum W, Warkentin B (eds) Function of soils for human societies and the environment. Special Publication of the Geological Society 266:89–115
Jiao H, Chen YL, Lin XG, Liu RJ (2011) Diversity of arbuscular mycorrhizal fungi in greenhouse soils continuously planted to watermelon in North China. Mycorrhiza 21:681–688
Johnson NC (1993) Can fertilizatioin of soil select less mutualistic mycorrhizae? Ecol Appl 3:749–757
Johnson D, Vandenkoornhuyse PJ, Leake JR, Gilbert L, Booth RE, Grime PJ, Young JPW, Read D (2003) Plant communities affect arbuscular mycorrhizal fungal diversity and community composition in grassland microcosms. New Phytol 161:503–515
Ju XT, Kou CL, Christie P, Dou ZX, Zhang FS (2007) Changes in the soil environment from excessive application of fertilizers and manures to two contrasting intensive cropping systems on the North China Plain. Environ Pollut 145:497–506
Kabir Z, O’Halloran IP, Widden P, Hamel C (1998) Vertical distribution of arbuscular mycorrhizal fungi under corn (Zea mays L) in no-till and conventional tillage systems. Mycorrhiza 8:53–55
Kiers ET, West SA, Denison RF (2002) Mediating mutualisms: farm management practices and evolutionary changes in symbiont co-operation. J Appl Ecol 39:745–754
Köhl L, Oehl F, van der Heijden MGA (2014) Agricultural practices indirectly influence plant productivity and ecosystem services through effects on soil biota. Ecol Appl 24:1842–1853
Krüger M, Stockinger H, Krüger C, Schüßler A (2009) DNA-based species level detection of Glomeromycota: one PCR primer set for all arbuscular mycorrhizal fungi. New Phytol 183:212–223
Kurle JE, Pfleger FL (1994) Arbuscular mycorrhizal fungus spore populations respond to conversions between low-input and conventional management practices in a corn–soybean rotation. J Agric 86:467–475
Lauber CL, Strickland MS, Bradford MA, Fierer N (2008) The influence of soil properties on the structure of bacterial and fungal communities across land use types. Soil Biol Biochem 40:2407–2415
Lekberg Y, Koide R, Twomlow S (2008) Effect of agricultural management practices on arbuscular mycorrhizal fungal abundance in low-input cropping systems of southern Africa: a case study from Zimbabwe. Biol Fert Soils 44:917–923
Li MG (2000) Operating principles and techniques of plant genes. Tianjin Science and Technology Press, Tianjin
Li XL, George E, Marschner H (1991a) Phosphorus depletion and pH decrease at the root-soil and hyphae–soil interfaces of VA-mycorrhizal white clover fertilized with ammonium. New Phytol 119:397–404
Li XL, Marschner H, George E (1991b) Acquisition of phosphorus and copper by VA-mycorrhizal hyphae and root to shoot transport in white clover. Plant Soil 136:49–57
Li XL, Gai JP, Cai XB, Li XL, Christie P, Zhang FS, Zhang JL (2014) Molecular diversity of arbuscular mycorrhizal fungi associated with two co-occurring perennial plant species on a Tibetan altitudinal gradient. Mycorrhiza 24(2):95–107
Liu RJ, Li M, Wang FY (2001) A preliminary research on the species diversity of arbuscular mycorrhizal fungi in the greenhouse vegetable lands. J Laiyang Agric Coll 18:280–283 (in Chinese)
Maherali H, Klironomos JN (2012) Phylogenetic and trait-based assembly of arbuscular mycorrhizal fungal communities. PLoS ONE 7:36695
Martinez TN, Johnson NC (2010) Agricultural management influences propagule densities and functioning of arbuscular mycorrhizas in low- and high-input agroecosystems in arid environments. Appl Soil Ecol 46:300–306
Meharg AA, Cairney JWG (2000) Co-evolution of mycorrhizal symbionts and their hosts to metal-contaminated environments. Adv Ecol Res 30:69–112
Moorman T, Reeves FB (1979) The role of endomycorrhizae in revegetation practices in the semi-arid west. Part II. A bioassay to determine the effect of land disturbance on endomycorrhizal populations. Am J Bot 66:14–18
Murty D, Kirschbaum MUF, McMurtrie RE, McGilvray A (2002) Does conversion of forest to agricultural land change soil carbon and nitrogen? A review of the literature. Glob Change Biol 8:105–123
Neville J, Tessier JL, Morrison I, Scarratt J, Canning B, Klironomos JN (2002) Soil depth distribution of ecto- and arbuscular mycorrhizal fungi associated with Populus tremuloides within a 3-year-old boreal forest clear-cut. Appl Soil Ecol 19:209–216
Newman EI, Reddell P (1987) The distribution of mycorrhizas among families of vascular plants. New Phytol 106:745–751
Oehl F, Sieverding E, Ineichen K, Mader P, Boller T, Wiemken A (2003) Impact of land use intensity on the species diversity of arbuscular mycorrhizal fungi in agroecosystems of central Europe. Appl Environ Microbiol 69:2816–2824
Oehl F, Sieverding E, Mader P, Dubois D, Ineichen K, Boller T, Wiemken A (2004) Impact of long-term conventional and organic farming on the diversity of arbuscular mycorrhizal fungi. Oecologia 138:547–583
Oehl F, Sieverding E, Ineichen K, Ris EA, Boller T, Wiemken A (2005) Community structure of arbuscular mycorrhizal fungi at different soil depths in extensively and intensively managed agroecosystems. New Phytol 165:273–283
Oehl F, Laczko E, Bogenrieder A, Stahr K, Bösch R, van der Heijden M, Sieverding E (2010) Soil type and land use intensity determine the composition of arbuscular mycorrhizal fungal communities. Soil Biol Biochem 42:724–738
Olsen SR, Sommers LE (1982) Phosphorus. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2. Am Soc Agron and Soil Sci Soc Am, Madison
Post WM, Mann LK (1990) Changes in soil organic carbon and nitrogen as a result of cultivation. In: Bowman AF (ed) Soil and the greenhouse effect. Wiley, New York, pp 401–407
Pozo MJ, Azcón-Aguilar C (2007) Unraveling mycorrhiza-induced resistance. Curr Opin Plant Biol 10:393–398
Qiu SJ, Ju XT, Ingwersen J, Qin ZC, Li L, Streck T, Christie P, Zhang FS (2010) Changes in soil carbon and nitrogen pools after shifting from conventional cereal to greenhouse vegetable production. Soil Tillage Res 107:80–87
Redecker D (2002) Molecular identification and phylogeny of arbuscular mycorrhizal fungi. Plant Soil 244:67–73
Redecker D, Hijri I, Wiemken A (2003) Molecular identification of arbuscular mycorrhizal fungi in roots: perspectives and problems. Folia Geobot 38:113–124
Richter BS, Tiller RL, Stutz JC (2002) Assessment of arbuscular mycorrhizal fungal propagules and colonization from abandoned agricultural fields and semi-arid grasslands in riparian floodplains. Appl Soil Ecol 20:227–238
Sanders IR (2004) Plant and arbuscular mycorrhizal fungal diversity—are we looking at the relevant levels of diversity and are we using the right techniques? New Phytol 164:415–418
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Simon L, Bousquet J, Lévesque RC, Lalonde M (1993) Origin and diversification of endomycorrhizal fungi and coincidence with vascular land plants. Nature 363:67–69
Smith SE, Read DJ (2008) Mycorrhizal symbiosis, 3rd edn. Academic, New York, London, p 787
Tawaraya K, Saito M, Morioka M, Waqatsuma T (1994) Effect of phosphate application to arbuscular mycorrhizal onion on the development and succinate dehydrogenase activity of internal hyphae. Soil Sci Plant Nutr 40:667–673
Vandenkoornhuyse P, Husband R, Daniell TJ, Watson IJ, Duck JM, Fitter AH, Young JPW (2002) Arbuscular mycorrhizal community composition associated with two plant species in a grassland ecosystem. Mol Ecol 11:1555–1564
Verbruggen E, Roling WFM, Gamper HA, Kowalchuk GA, Verhoef HA, vander Heijden MGA (2010) Positive effects of organic farming on below-ground mutualists: large-scale comparison of mycorrhizal fungal communities in agricultural soils. New Phytol 186:968–979
Zhu JH, Li XL, Christie P, Li JL (2005) Environmental implications of low nitrogen use efficiency in excessively fertilized hot pepper (Capsicum frutescens L.) cropping systems. Agric Ecosyst Environ 111:70–80
Acknowledgments
We thank the National Basic Research Program of China (Project 2009CB119000), the innovative group grant of NSFC (No. 31121062), and the National Natural Science Foundation of China (Projects 41071179 and 41271269) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Ian Anderson
Rights and permissions
About this article

Cite this article
Gai, J., Gao, W., Liu, L. et al. Infectivity and community composition of arbuscular mycorrhizal fungi from different soil depths in intensively managed agricultural ecosystems. J Soils Sediments 15, 1200–1211 (2015). https://doi.org/10.1007/s11368-015-1060-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-015-1060-3