
Abstract
Purpose
Iron’s fluctuation between the II (ferrous) and III (ferric) oxidation states has been coined as the “FeIII–FeII redox wheel.” Numerous studies have coupled the “iron redox wheel” with the biogeochemical cycle of carbon (C), nitrogen (N), sulfur (S), or phosphorus (P) individually in soils or sediments, but evidence suggests that the FeIII–FeII redox wheel drives the biogeochemical cycles interactively in a fluctuating redox microenvironment. The interactions of the FeIII–FeII redox wheel with the biogeochemical cycles of C, N, S, and P in the fluctuating redox environments were reviewed in this paper.
Discussion
In this review, we discuss the importance of iron with regard to each of the biogeochemical cycles individually as well as interactively. The importance of crystalline and non-crystalline FeIII (hydr)oxides is highlighted as they serve as terminal electron acceptors for organic matter mineralization and N and S transformation and also act as sorbents for dissolved P compounds. Mechanically, electron transfer from organic matter to FeIII (hydr)oxides via organic matter oxidation, oxidation of NH +4 to NO −2 , formation and oxidation of Fe sulfide minerals in the S cycle, and P transformation were discussed to couple with the FeIII–FeII redox wheel.
Conclusions
The knowledge gaps are identified at the end of the review. The natural environmental relationships still require further studies that link the iron redox wheel as a driver of the biogeochemical cycles of C, N, S, and P. Anthropogenically altered environments (nutrient and metal elevation, global warming, and acidification) require intensive studies to allow for improved integrated modeling of global C, N, S, and P biogeochemical cycles driven by the FeIII–FeII redox wheel.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The iron (Fe) redox cycle refers ferrous iron (FeII) oxidation and ferric iron (FeIII) reduction, referred to as the “FeIII–FeII redox wheel” in this review, which is driven by ambient E h/pH and microbial activity in a natural environment. Ferric iron (FeIII) is soluble under arguably acidic conditions (pH < 3 or <5), but it exists as FeIII (hydr)oxides at near-neutral pH (Lindsay 1979). Reduction of FeIII or FeIII (hydr)oxides to FeII occurs via either the abiotic or biotic pathway in a soluble, adsorbed, or solid state. The mechanisms and processes of the reductive dissolution of FeIII (hydr)oxides have been reviewed from both abiotic (Stumm and Sulzberger 1992) and biotic (Lovley et al. 2004) standpoints. In a reducing environment, FeIII (hydr)oxides may be reduced abiotically via surface processes (Stumm and Sulzberger 1992; Tanwar et al. 2009; Catalano et al. 2010). Alternatively, the reduction of FeIII (hydr)oxides can be driven by dissimilatory iron-reducing bacteria, coupling with the oxidation of organic carbon or H2 (Lovley et al. 2004; Lovley 2008). The FeII compounds, a group of strong reductants, can reduce FeIII (hydr)oxides and form secondary minerals such as goethite, siderite, magnetite, and green rusts (Borch et al. 2010). The oxidation of FeII may occur when phyto-photosynthesis results in higher ambient O2 concentrations or when mediated by anoxygenic photosynthetic bacteria under low O2 concentrations (Kappler and Straub 2005).
Numeric research has pointed out that the FeIII–FeII redox wheel couples with biogeochemical cycles of carbon (C), nitrogen (N), sulfur (S), or phosphorus (P) individually or interactively. The FeIII–FeII redox wheel not only acts as an electron donor and acceptor coupling with the reduction–oxidation of C, N, and S but also provides adsorption surface such as secondary iron minerals, especially for P. In this review, we present an overview of the interactions of the FeIII–FeII redox wheel with the C, N, S, and P biogeochemical cycles occurring in soils and sediments. We highlight recent developments and the most pressing needs in this field.
2 The roles of the FeIII–FeII redox wheel within the biogeochemical cycles
2.1 Carbon cycling
A large reservoir of organic matter exists in reductive soils and sediments. This represents a thermodynamically unstable carbon source as it is susceptible to microbial decomposition. The microbial transformation releases protons and electrons, which are normally depleted through redox processes by electron acceptors (oxidants) such as O2, NO −3 , FeIII, SO 2−4 , and CO2 (Lindsay 1979; Bohn 1985). This may be illustrated with an example:
(Lindsay 1979).
Sahrawat (2004a) pointed out the lack of FeIII (hydr)oxides as terminal electron acceptors increases organic matter accumulation in submerged soils and sediments (Sahrawat 2004a). Roden and Wetzal (2002) confirmed that the first-order constant of FeIII reduction was correlated to the initial mineralization rate (as ∑CO2 + CH4) of organic matter in freshwater sediments. Thus, the FeIII–FeII redox wheel is considered an important redox process that links the reduction of FeIII (hydr)oxides to the oxidation (or mineralization) of organic matter.
Organic ligands may be produced via the anaerobic decomposition of organic matter when a soil or sedimentary environment becomes reductive (Sahrawat 2004a). In the presence of these organic ligands, the dissolution of FeIII (hydr)oxides is thermodynamically controlled by surface processes as opposed to mass transport processes (Stumm and Sulzberger 1992). The formation of complexes (or chelates) between organic ligands (such as oxalate) with FeIII on the FeIII (hydr)oxides’ surface can polarize the FeIII–O bonds, which weaken or even break the bonds and enhance the release of FeIII into the solution as organic FeIII complexes (Ratering and Schnell 2000). Alternatively, reactive functional groups of these organic ligands (such as hydroxyl and carboxyl groups) may complex with the FeIII (hydr)oxide surface and transfer their inner-spherical electrons and reduce FeIII (hydr)oxides interstructurally. The FeII–O bonds of the reduced crystalline lattice are easily broken and release FeII into the solution (Larsen et al. 2006; Hofmann and Liang 2007; Debnath et al. 2010). On the other hand, soluble organic ligand-bound FeII can attack FeIII (hydr)oxides, resulting in FeIII (hydr)oxide dissolution (Suter et al. 1991). Therefore, the induced reductive dissolution of FeIII (hydr)oxides can be summarized via three steps: (1) rapid adsorption or diffusion of reductive organic ligand to the oxides’ surface; (2) the transfer of the inner-spherical electrons to the oxides’ surface (reduction of FeIII to FeII); and (3) the detachment and diffusion of FeII into the bulk solution (Stumm and Sulzberger 1992). The dissolution rate depends on the specific surface area and structure of the FeIII (hydr)oxides. Another factor is the presence of FeII on the surface as this may block the active sites of the FeIII (hydr)oxides and lower the reductive dissolution rate. Additionally, the dissolution rate is pH-dependent as the free energy of the electron transfer reaction decreases as the environment becomes more alkaline (Stumm and Sulzberger 1992).
Biologically, the microorganisms can enzymatically couple the oxidation of organic matter to the reduction of FeIII (hydr)oxides (Christen 2001; Roden and Wetzal 2002; Lovley et al. 2004; Beal et al. 2009; Hori et al. 2010). Lovley (1995) constructed a model where the oxidation of organic matter to CO2 occurred with FeIII acting as the sole electron acceptor (Fig. 1). In this model, monomeric compounds (such as sugars and amino acids), long-chain fatty acids, and mono-aromatics were hydrolytically released by microbial enzymes. These compounds were microbially hydrolysed to short-chain fatty acids and H2, which were further oxidized to CO2 and H2O by FeIII-reducing microorganisms.

Model for the microbial oxidation of organic matter in anaerobic soil/sediment with FeIII serving as the electron acceptor (Lovley 1995). In this model, complex organic matter is enzymatically hydrolyzed to be sugars, amino acids, long-chain fatty acids, and aromatics. Furthermore, fermentative microorganisms ferment sugars and amino acids to be short-chain fatty acids and H2. The fermentative products are then oxidized with the reduction of FeIII. The long-chain fatty acids and aromatics are also oxidized to CO2 and H2O by FeIII reducers. *SCFA short-chain fatty acid
Humic substances may function as an electron shuttle between FeIII-reducing microorganisms and insoluble FeIII (hydr)oxides, thereby coupling the oxidation of organic matter to the reduction of FeIII (hydr)oxides in reductive soils or sediments (Lovley et al. 1996; Kappler et al. 2004; Jiang and Kappler 2008; Rakshit et al. 2009). Microorganisms may also reduce FeIII through direct (Lovley et al. 1991; Lovley 1995) or indirect (Lovley et al. 1998) physical contact with insoluble FeIII (hydr)oxides in the presence of dissolved humic substances. This contradicted the paradigm that suggested that humic substances derived from the decomposition of soil organic matter were inert and resisted decay in the soil for many years (Bohn 1985).
Quinone moieties act as the predominant electron acceptors within humic substances, and their reduced forms can transfer electrons to either dissolved or solid phase FeIII (Scott et al. 1998; Nurmi and Trathyek 2002; Cory and McKnight 2005; Aeschbacher et al. 2010). The microbial electron transfer is dependent on the concentration of the humic substances present. Electron shuttling via humic substances to ferrihydrite would not occur in aqueous environments where dissolved humic substances occurred below a threshold of 5 mg C per liter (Jiang and Kappler 2008). The electron-carrying capacity depends upon the chemical structure of humic substances and the environmental conditions under which the humic substances are produced (Lovley and Blunt-Harris 1999; Struyk and Sposito 2001; Bauer et al. 2007). Dissolved organic matter (DOM) acts as the most viable electron shuttle in sediments and aquatic ecosystems, partly because of its ubiquity and partly due to its quinone functional groups and metal-chelating moieties (Hakala and Weber 2007; Fimmen et al. 2007; Zhang and Weber 2009).
In summary, the FeIII–FeII redox wheel is interactively coupled with C cycling in a reductive environment. The integration of organic matter mineralization and the FeIII–FeII redox wheel may also play an important role in the concurrent cycling of N, S, and P since organic matter mineralization is a crucial process for N, S, and P cycles.
2.2 Nitrogen cycling
The excessive N in the environment resulting from anthropogenic activity (fertilizer use and fossil fuel combustion) increases eutrophication, acid rain, and harmful algal blooms (Seitzinger 2008; Schlesinger 2009). Soils are the dominant N sink in an ecosystem, although their N content may be lowered by plant uptake (Nadelhoffer et al. 1999). Intensive research pertaining to the transformation and retention of N in soils and sediments has increased the understanding of N cycling in terrestrial and aquatic ecosystems. The FeIII–FeII redox wheel has been abiotically and biotically involved within the N cycling. Microbially mediated oxidation of FeII coupling nitrate (NO −3 ) reduction has been reported within a variety of natural wetland soils and sediments (Senn and Hemond 2002; Straub et al. 2004; Weber et al. 2006; Smolders et al. 2010). The coupling process was detected via enrichment culture and the most probable number method (Straub and Buchholz-Cleven 1998). Sideroxydans is identified as the primary genus responsible for lithoautotrophic FeII oxidation in a lithoautotrophic NO −3 -reducing enrichment culture (Straub et al. 1996; Blöthe and Roden 2009). However, its biochemical mechanisms and the genes responsible for the NO −3 -dependent FeII oxidation remain unclear (Blöthe and Roden 2009; Byrne-Bailey et al. 2010). Furthermore, whether the NO −3 -dependent FeII oxidation and FeIII (hydr)oxide reduction can concurrently occur in a reductive soil or sediment remains unknown, even though the redox condition for both processes should be similar. If they did occur coupledly, the FeIII–FeII redox wheel could be proven as involving both C and N cycles.
The reduction of NO −3 coupled to FeII oxidation may also proceed via abiotic pathways. The reduction of NO −3 (electron acceptor) to NO −2 with the concomitant oxidation of FeII (electron donor) to FeIII is thermodynamically favorable (Ottley et al. 1997). Davidson et al. (2003) proposed the “ferrous wheel hypothesis” that FeIII (hydr)oxide reduction by the carbonaceous compounds is followed by NO −3 to nitrite (NO −2 ) reduction and FeII to FeIII oxidation. The NO −2 subsequently reacted with DOM to produce dissolved organic nitrogen complexes (Fig. 2). This was hypothesized to occur in an anaerobic microsite that contained FeII and an excess of organic carbon (Davidson et al. 2003, 2008). The reaction of NO −2 with DOM was based upon the rapid reaction of NO −2 with phenolic compounds (Davidson et al. 2003) instead of a rapid incorporation of NO −2 into solid organic matter (Thorn and Mikita 2000; Fitzhugh et al. 2003). The hypothesis could explain the rapid disappearance of NO −3 in soils and sediments (Corre et al. 2007; Burgin and Hamilton 2007; Zhu and Wang 2011). This is an evidence of the FeIII–FeII redox wheel interacting with C and N biochemical cycles.

Conceptual model illustrating relationships between the FeIII–FeII redox wheel and N cycling in anaerobic soil/sediment. In this model, the “ferrous wheel hypothesis” established by Davidson et al. (2003) shows that carbon compounds derived from photosynthates reduce FeIII in soil minerals and FeII is released into the solution. FeII further reduces NO −3 to NO −2 , and NO −2 subsequently reacts with DOM to produce DON in temperate forest soils under anaerobic condition. In addition, the reduction of FeIII in minerals, such as FeIII (hydr)oxides and octahedral FeIII, affects their surface structure and negatively charges solid phases, which induces NH +4 movement between aqueous and solid phases in wetland and paddy soils. The production of NO −2 and FeII may also simultaneously occur in wetland soils from the coupled reduction of FeIII to FeII with the oxidation of NH +4 to NO −2 . *DOM dissolved organic matter, **DON dissolved organic nitrogen
However, the rapid abiotic reaction of NO −3 with FeII in the ferrous wheel hypothesis was questioned by Colman et al. (2007, 2008). They failed to repeat the catalytic reduction of NO −3 to produce NO −2 instead of NH3 or N2 as the end product in 45 soils. They concluded that the catalytic reduction of NO −3 would not occur in acidic forest soils. Interestingly, an unlikely abiotic reaction of NO −2 with natural dissolved organic carbon was reported by Schmidt and Matzner (2009). Debates on the ferrous wheel hypothesis are ongoing, but evidences from both sides support our hypothesis that the FeIII–FeII redox wheel has been involved in the cycling of C and N interactively in the redox fluctuating soil or sediment systems.
The role of FeIII has been demonstrated with regard to N mineralization or ammonium (NH +4 ) production in anaerobic environments (see Fig. 2). Organic matter and FeIII are two important factors that control NH +4 production in submerged soils or sediments (Sahrawat 2004b; Coyle et al. 2009; Yin et al. 2010) and have been correlated with NH +4 production in paddy soils (Sahrawat and Narteh 2003). Zhang and Scherer (1999, 2000) demonstrated that the diffusion of NH +4 into or out of clay was promoted by the reductive dissolution of FeIII (hydr)oxides which coated the clay particles. On the other hand, flooding enhances the reduction of octahedral FeIII and negative charges within layers of clay minerals. These two enhanced characteristics may accelerate the diffusion of NH +4 into clay and its subsequent fixation in paddy soils (Scherer and Zhang 2002; Liu et al. 2008; Nieder et al. 2011).
Additionally, simultaneous production of NO −2 and FeII may occur under strict anaerobic conditions in soil slurries (Clément et al. 2005; Burgin et al. 2011) via the coupled reduction of FeIII to FeII with the subsequent oxidation of NH +4 to NO −2 (see Fig. 2). The relative importance of abiotic or biotic processes involved in the coupled oxidation of NH +4 and reduction of FeIII, as well as their ecological effects under field conditions, still requires further elucidation. However, studies indicated that N cycling in dynamic redox environments is related to C cycling and the FeIII–FeII redox wheel abiotically and biotically.
2.3 Sulfur cycling
Iron sulfides couple S cycling with the FeIII–FeII redox wheel. This has been supported by the processes and fluxes occurring in the global geochemical iron sulfide cycle and the “iron–sulfur world” hypothesis (Rickard and Luther 2007). The chemistry of SII suggests that H2S and HS− are two main species in pore water of marine sediment profiles while S2− is negligible (Rickard and Luther 2007). Aqueous iron sulfide–complexes produced by the reaction of H2S or HS− with FeII are prevalent in the literature (Fig. 3). Based on the stoichiometry, soluble iron–sulfide complexes, such as Fe2S2 (Buffle et al. 1988), Fe x (HS)2x (x ≥ 2) (Davison et al. 1998), and [Fe2(HS)]3+ (Luther et al. 2003), have been suggested, while some authors refer to the soluble iron–sulfide complexes as FeSaq (Rickard and Morse 2005) with no implication of stoichiometry. Given the above, soluble iron–sulfide complexes may occur as aqueous FeS clusters, and their apparent stability is likely to be an average for the cluster system. More importantly, FeSaq plays a key role in the geochemical dynamics of ecosystems as it removes toxic H2S and drives hydrothermal vent ecosystems (Luther et al. 2001; Luther and Rickard 2005). FeSaq is also a necessary intermediate in the formation of solid Fe sulfides, such as mackinawite and pyrite (Luther et al. 2003; Slowey and Brown 2007; Papadas et al. 2009). Oxidation of FeSaq on FeIII (hydr)oxides’ surface releases FeII and SO 2−4 into the solution. Soluble FeII further reacts with another FeSaq to form Fe sulfide minerals. Thus, S transformation between aqueous and solid phases is controlled by FeIII (hydr)oxides (Poulton et al. 2004; Zopfi et al. 2008).

Conceptual model illustrating the relationships between the FeIII–FeII redox wheel and S cycling in anaerobic soil/sediment. In this model, the reduction of FeIII to FeII couples with the transformation of S2O 2−3 to S4O 2−6 and S 08 to SO 2−4 , while the oxidation of FeII to FeIII couples with the transformation of SO 2−4 to HS− and H2S. The formation of soluble FeS and FeS2 also regulates the transformation of HS− and H2S to S 08 and S2O 2−3 , respectively
The Fe sulfide minerals are the major sinks of reduced S in anaerobic subsurface environments (Rickard and Morse 2005). The formation of Fe sulfide minerals promotes the immobilization of dissolved FeII as well as toxic trace elements (Kirk et al. 2010; Johnston et al. 2010). Pyrite is one of the major iron sulfide minerals for sequestrating reduced S (Rickard and Morse 2005). Pyrite may form with (Hunger and Benning 2007) or without (Burton et al. 2011) the need of FeS precursors, such as mackinawite (FeS) and greigite (Fe3O4). This potentially causes a spatial decoupling between monosulfide accumulation and pyrite stability (Burton et al. 2011), indicating the complexity of Fe–S mineralization pathways in a reductive environment.
Iron sulfide minerals are likely to be oxidized when reductive soils or sediments are disturbed or resuspended into an oxic environment. The oxidation of Fe sulfide minerals may be coupled to FeIII (hydr)oxide reduction (Zopfi et al. 2008; Pollok et al. 2009; Aller et al. 2010; Li et al. 2011), resulting in acidification (Boman et al. 2010; Claff et al. 2011; Keene et al. 2011) and releasing dissolved FeII and toxic trace elements (Kocar et al. 2010; Burton et al. 2011). A variety of key intermediate S compounds were observed (Burton et al. 2009; Zopfi et al. 2008) during the oxidation of Fe sulfide minerals (see Fig. 3), including elemental S (S 08 ), thiosulfate (S2O 2−3 ), trithionate (S3O 2−6 ), tetrathionate (S4O 2−6 ), and disulfane-monosulfonic acid (HS3O −3 ).
In short, the FeIII–FeII redox wheel not only relates to Fe–S mineralization but also is involved in the oxidation of Fe sulfides. Interestingly, CO2/CO can be reduced by FeSaq to form organic sulfides under extreme environments (Rickard and Luther 2007). It suggests that C and S cycling interacts with the FeIII–FeII redox wheel.
2.4 Phosphorus cycling
The transformation of P in soils or sediments has become a significant eutrophication risk as it may be released into adjacent aquatic ecosystems (Carpenter 2008; Howarth and Paerl 2008; Conley et al. 2009; Duan et al. 2009). Phosphorus has a strong affinity toward FeIII (hydr)oxide surfaces (Khare et al. 2004; Norton et al. 2009). Since FeIII (hydr)oxide is redox-sensitive and its surface is variably charged, P sorption on the FeIII (hydr)oxide surface becomes dependent on the redox potential and pH of the environment. The transformation and distribution of P compounds from the aqueous phase to solid phase iron oxide minerals depends on the type of iron oxide and its surface structure (Fig. 4). The reactive surface areas of amorphous FeII or FeIII (hydr)oxides are much greater than those of crystalline FeIII (hydr)oxides. Amorphous FeIII (hydr)oxides can enhance the sorption of dissolved inorganic phosphorus (DIP) in a reductive soil (Zhang et al. 2010). The co-precipitation of P and FeIII (hydr)oxides due to oxidation (via nitrate reduction or aeration) leads to a high retention of P in non-calcareous sediments (Schauser et al. 2006; Lake et al. 2007). Vivianite (Fe3(PO4)2⋅8H2O) or similar FeII phosphate minerals enhance the P sorption capacity in reductive mineral soils with high DIP (Heiberg et al. 2010). Dissolved organic phosphorus adsorption displayed a similar trend, with less sorption occurring as the crystallinity of FeIII (hydr)oxides increased (Ruttenberg and Sulak 2011).

Conceptual model illustrating the relationships between the FeIII–FeII redox wheel and P cycling in anaerobic soil/sediment. In this model, sorption on the surface of amorphous FeII or FeIII (hydr)oxides, precipitation as FeII phosphate, and co-precipitation with FeIII (hydr)oxides promote the transformation of P from the soluble to the solid phase. In contrast, reductive dissolution of FeIII (hydr)oxides decreases P-binding sites and increases negative charges on solid phase surface, inducing the transformation of P from the solid to the soluble phase. The formation of Fe sulfide minerals in sulfate-enriched anaerobic sediments can promote P release by preventing the formation of insoluble Fe–phosphate complexes or by hampering FeII diffusion to aerobic layers. Soluble Fe–humic complexes may also accelerate P release from the solid to the soluble phase via blocking P co-precipitating with FeIII (hydr)oxides
Nonetheless, the release of P from Fe–P complexes is coupled with the FeIII–FeII redox wheel in the environment (see Fig. 4). The reductive dissolution of FeIII (hydr)oxides at alkaline pHs has been correlated to greater P release in submerged mineral soils or sediments (Chacon et al. 2006; Mort et al. 2010; Heiberg et al. 2010). However, the presence of aluminum hydroxides (Al(OH)3) lowered the P release in the aqueous phase of non-calcareous sediments—even though an in situ reductive solubilization of FeIII (hydr)oxides occurred (Kopáček et al. 2005). Aluminum hydroxides are not sensitive to redox fluctuation, but pH-dependent. Aluminum hydroxides may provide additional binding sites for P if P is released from FeIII (hydr)oxides at neutral pH (Kopáček et al. 2000; Rydin et al. 2000; Reitzel et al. 2005; Navratil et al. 2009). Kopáček et al. (2005) suggested that the molar Al/Fe ratio and the molar Al/P ratio were indicative parameters for predicting P release from lake sediments. This has been corroborated by Lake et al. (2007).
FeIII can form soluble complexes with humic (Steinmann and Shotyk 1997). The stable Fe–humic complexes resulted in P release in eutrophic fens since they prevent P from co-precipitating with FeIII (hydr)oxides (Zak et al. 2004). The formation of Fe–S complexes in sulfate-enriched anaerobic sediments can also promote P release by preventing the formation of insoluble Fe–phosphate complexes or by hampering FeII diffusion to aerobic layers (Smolders et al. 2010; Hartzell and Jordan 2012). The Fe/P ratio may be used to predict P sorption capacity for a sediment. A significantly negative relationship was found between soluble P and the total Fe/P ratio in the trophogenic zone of surface sediments from Danish lakes (Jensen et al. 1992). Reductive dissolution of FeIII (hydr)oxides (Section 2.1) caused by anaerobic microbial respiration can enhance P solubilization as well (Pant and Reddy 2001; Zak et al. 2004; Shenker et al. 2005; Chacon et al. 2006; Zhang et al. 2010).
Overall, the FeIII–FeII redox wheel only regulates sorption surface or site for phosphorus in the environment regardless of Fe–humic complex formation or dissolution of Fe oxides. Phosphorus released from organic P mineralization coupled with FeIII reduction might relate to the FeIII–FeII redox wheel.
2.5 Integration of biogeochemical cycles with the FeIII–FeII redox wheel concept
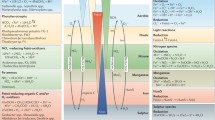
The FeIII–FeII redox wheel plays an important role in the biogeochemical cycles of C, N, S, and P, and at least partially drives or is involved into their biogeochemical cycles at various scales. It is evident that FeIII (hydr)oxides are the core for the biogeochemical cycles of C, N, S, and P in a dynamic redox micro-ecosystem, where they serve as terminal electron acceptors for organic matter mineralization and N and S transformation. They also possess a remarkably large specific surface area and act as sorbents for dissolved P compounds. With the above factors in mind, the C, N, S and P cycles were integrated with the FeIII–FeII redox wheel concept; the conceptual model is displayed in Fig. 5. In the model, FeIII serves as terminal electron acceptors for organic matter mineralization; the reduction of FeIII to FeII couples the transformation of NO −3 to NO −2 , NH +4 to NO −2 , S2O 2−3 to S4O 2−6 , and S 08 to SO 2−4 ; the oxidation of FeII to FeIII couples the transformation of SO 2−4 to HS− and H2S; the Fe redox processes change the surface structure of the solid phase and the solution chemistry, affecting the transformation of NH +4 and P between soluble and solid phases. Some evidences also indicate that the FeIII–FeII redox wheel integrates with the C and N, C and S, or C and P cycles interactively, especially the C and N cycles (Davidson et al. 2003; Colman et al. 2007), in the dynamic redox environments.

Conceptual model illustrating the relationships between the FeIII–FeII redox wheel and C, N, S, and P cycles in anaerobic soil/sediment. In this model, the FeIII–FeII redox wheel drives the biogeochemical cycles of C, N, S, and P interactively in dynamic redox environments. FeIII serves as a terminal electron acceptor for organic matter mineralization. The reduction of FeIII to FeII drives the transformation of NO −3 to NO −2 , NH +4 to NO −2 , S2O 2−3 to S4O 2−6 , and S 08 to SO 2−4 , while the oxidation of FeII to FeIII couples the transformation of SO 2−4 to HS− and H2S. Furthermore, the FeIII–FeII redox wheel changes the surface structure of solid phase and solution chemistry, affecting the transformation of NH +4 and P between the soluble and solid phases in anaerobic soil/sediment
Although iron is one of the most abundant elements in the earth’s crust, it may be considered as limited within a microsite or micro-ecosystem for reduction or oxidation. When iron is chemically inert (due to Fe–S precipitation or Fe–humic complexation), FeIII (hydr)oxides are reduced (via abiotic or biotic nitrification processes), or the specific surface area of FeIII (hydr)oxides decreases (due to crystallization), the P in the aqueous phase of the micro-ecosystem may increase due to the lack of a sorption surface and the co-precipitation of FeIII oxide minerals. The reverse is true that the availability of C, S or N may be lowered due to the oxidation or complexation of Fe oxides. Given the complexity of the interactive processes between the FeIII–FeII redox wheel and C, N, S, and P biogeochemical cycles and the difficulty of locating certain dynamic redox environment, direct evidences are demanded in future studies.
3 Challenges and future studies
There is still a great need to understand the interactions of biogeochemical C, N, S, and P cycles with the FeIII–FeII redox wheel in microenvironments under aerobic–anaerobic fluctuations, even though the complexity of coupling these relationships has been discussed in previous studies (Gauci et al. 2004; Robinson et al. 2004; Meulepas et al. 2009; Howarth et al. 2011; Burgin et al. 2011; Schlesinger et al. 2011). Nowadays, anthropogenic activities result in nutrient and metal enrichment in the environment, increase ambient temperature, and alter pH in environmental matrices. This, in turn, alters the interactions of the biogeochemical cycles and increases the complexity of predicting their interactions with the FeIII–FeII redox wheel. However, these challenges must be overcome because they are crucial to process-based models that predict these cycles in anthropogenically altered ecosystems. Despite some advances, more work should be focused on the interactions of the FeIII–FeII redox wheel involved in the cycles of C, N, S, and P. The major challenges are stated below:
-
1.
Studies in anaerobic environments have demonstrated the significant role of humic substances on electron transfer from many microorganisms to crystalline and non-crystalline FeIII (hydr)oxides (Section 2.1). Nonetheless, direct evidence of kinetic mechanisms of electron transfer is lacking.
-
2.
The “ferrous wheel hypothesis” proposed by Davidson et al. (2003) requires confirmation or further refinement due to some contradictory findings (Section 2.2). The natural dissolved organic carbon extracted from different types of soils and sediments should be used to investigate the possibility of its abiotic reaction with NO −2 under anaerobic conditions. The rapid catalytic reduction of NO −3 mediated by FeII adsorbed to FeIII (hydr)oxide surfaces also requires confirmation.
-
3.
So far, FeII sulfides have been defined and quantified on the basis of the source of H2S evolved from soils and sediments by adding 1 mol l−1 HCl (Rickard and Morse 2005). Species of FeII sulfides and their presence in anaerobic environmental conditions require new in situ measurement techniques, as opposed to the operationally defined quantification (Section 2.3).
-
4.
Although some studies have assessed the movement and transformation of P, the mechanisms controlling the release or retention of P in anaerobic soils and sediments remain unknown. It is well documented that some physical and chemical factors (such as pH, redox potential, available P concentration, Al hydroxides, FeIII (hydr)oxides) will influence the adsorption–desorption processes of P in reducing soils or sediments (Section 2.4). But, the integrative mechanisms of these factors regulating P sorption or desorption in relation the FeIII–FeII redox wheel require direct evidence.
References
Aeschbacher M, Sander M, Schwarzenbach RP (2010) Novel electrochemical approach to assess the redox properties of humic substances. Environ Sci Technol 44:87–93
Aller RC, Madrid V, Chistoserdov A, Aller JY, Heilbrum C (2010) Unsteady diagenetic processes and sulfur biogeochemistry in tropical deltaic muds: implications for oceanic isotope cycles and the sedimentary record. Geochim Cosmochim Acta 74:4671–4692
Bauer M, Heitmann T, Macalady DL, Blodau C (2007) Electron transfer capacities and reaction kinetics of peat dissolved organic matter. Environ Sci Technol 41:139–145
Beal EJ, House CH, Orphan VJ (2009) Manganese- and iron-dependent marine methane oxidation. Science 325:184–187
Blöthe M, Roden EE (2009) Composition and activity of an autotrophic Fe(II)-oxidizing, nitrate-reducing enrichment culture. Appl Environ Microbiol 75:6937–6940
Bohn HL (1985) Oxidation and reduction. In: Bohn HL (ed) Soil chemistry. Wiley, New York, pp 262–289
Boman A, Fröjdö S, Backlund K, Åström ME (2010) Impact of isostatic land uplift and artificial drainage on oxidation of brackish-water sediments rich in metastable iron sulfide. Geochim Cosmochim Acta 74:1268–1281
Borch T, Kretzschmar R, Kappler A, Van Cappellen P, Ginder-vogel M, Voegelin A, Campbell K (2010) Biogeochemical redox processes and their impact on contaminant dynamics. Environ Sci Technol 44:15–23
Buffle J, de Vitre RR, Perret D, Leppard GG (1988) Combining field measurements for speciation in non perturbable waters. In: Kramer JR Jr, Allen HE (eds) Metal speciation: theory, analysis and application. Lewis Publishers, Boca Raton, pp 99–124
Burgin AJ, Hamilton SK (2007) Have we overemphasized the role of denitrification in aquatic ecosystems? A review of nitrate removal pathways. Front Ecol Environ 5:89–96
Burgin AJ, Yang WH, Hamilton SK, Silver WL (2011) Beyond carbon and nitrogen: how the microbial energy economy couples elemental cycles in diverse ecosystems. Front Ecol Environ 9:44–52
Burton ED, Bush RT, Sullivan LA, Hocking RK, Mitchell DRG, Johnston SG, Fitzpatrick RW, Raven M, Mcclure S, Jang LY (2009) Iron-monosulfide oxidation in natural sediments: resolving microbially mediated S transformations using XANES, electron microscopy, and selective extractions. Environ Sci Technol 43:3128–3134
Burton ED, Bush RT, Johnston SG, Sullivan LA, Keene AF (2011) Sulfur biogeochemical cycling and novel Fe–S mineralization pathways in a tidally re-flooded wetland. Geochim Cosmochim Acta 75:3434–3451
Byrne-Bailey KG, Weber KA, Chair AH, Bose S, Knox T, Spanbauer TL, Chertkov O, Coates JD (2010) Completed genome sequence of the anaerobic iron-oxidizing bacterium Acidovorax ebreus strain TPSY. J Bacteriol 192:1475–1476
Carpenter SR (2008) Phosphorus control is critical to mitigating eutrophication. Proc Natl Acad Sci 105:11039–11040
Catalano JG, Fenter P, Park C, Zhang Z, Rosso KM (2010) Structure and oxidation state of hematite surfaces reacted with aqueous Fe(II) at acidic and neutral pH. Geochim Cosmochim Acta 74:1498–1512
Chacon N, Silver WL, Dubinsky EA, Cusack DF (2006) Iron reduction and soil phosphorus solubilization in humid tropical forests soils: the roles of labile carbon pools and an electron shuttle compound. Biogeochemistry 78:67–84
Christen K (2001) Linking iron with carbon sequestration. Environ Sci Technol 35:98A–99A
Claff SR, Burton ED, Sullivan LA, Bush RT (2011) Metal partitioning dynamics during the oxidation and acidification of sulfidic soil. Chem Geol 286:146–157
Clément JC, Shrestha J, Ehrenfeld JG, Jaffé PR (2005) Ammonium oxidation coupled to dissimilatory reduction of iron under anaerobic conditions in wetland soils. Soil Biol Biochem 37:2323–2328
Colman BP, Fierer N, Schimel JP (2007) Abiotic nitrate incorporation in soil: is it real? Biogeochemistry 84:161–169
Colman BP, Fierer N, Schimel JP (2008) Abiotic nitrate incorporation, anaerobic microsites, and the ferrous wheel. Biogeochemistry 91:223–227
Conley DJ, Paerl HW, Howarth RW, Boesch DF, Seitzinger SP, Havens KE, Lancelot C, Likens GE (2009) Controlling eutrophication: nitrogen and phosphorus. Science 323:1014–1015
Corre MD, Brumme R, Veldkamp E, Beese FO (2007) Changes in nitrogen cycling and retention processes in soils under spruce forests along a nitrogen enrichment gradient in Germany. Glob Chang Biol 13:1509–1527
Cory RM, McKnight DM (2005) Fluorescence spectroscopy reveals ubiquitous presence of oxidized and reduced quinones in dissolved organic matter. Environ Sci Technol 39:8142–8149
Coyle JS, Dijkstra P, Doucett RR, Schwartz E, Hart SC, Hungate BA (2009) Relationships between C and N availability, substrate age, and natural abundance 13C and 15N signatures of soil microbial biomass in a semiarid climate. Soil Biol Biochem 41:1605–1611
Davidson EA, Chorover J, Bryan Dail D (2003) A mechanism of abiotic immobilization of nitrate in forest ecosystems: the ferrous wheel hypothesis. Glob Chang Biol 9:228–236
Davidson EA, Dail DB, Chorover J (2008) Iron interference in the quantification of nitrate in soil extracts and its effect on hypothesized abiotic immobilization of nitrate. Biogeochemistry 90:65–73
Davison W, Phillips N, Tabner BJ (1998) Soluble iron sulfide species in natural waters: reappraisal of their stoichiometry and stability constants. Aquat Sci 377:193–203
Debnath S, Hausner DB, Strongin DR, Kubicki J (2010) Reductive dissolution of ferrihydrite by ascorbic acid and the inhibiting effect of phospholipid. J Colloid Interface Sci 341:215–223
Duan H, Ma R, Xu X, Kong F, Zhang S, Kong W, Hao J, Shang L (2009) Two-decade reconstruction of algal blooms in China’s lake Taihu. Environ Sci Technol 43:3522–3528
Fimmen RL, Cory RM, Chin YP, Trouts TD, McKnight DM (2007) Probing the oxidation–reduction properties of terrestrially and microbially derived dissolved organic matter. Geochim Cosmochim Acta 71:3003–3015
Fitzhugh RD, Lovett G, Venterea RT (2003) Biotic and abiotic immobilization of ammonium, nitrite, and nitrate in soils developed under different tree species in the Catskill Mountains, New York, USA. Glob Chang Biol 9:1591–1601
Gauci V, Matthews E, Dise N, Walter B, Koch D, Granberg G, Vile M (2004) Sulfur pollution suppression of the wetland methane source in the 20th and 21st centuries. Proc Natl Acad Sci 101:12583–12587
Hakala JACY, Weber EJ (2007) Influence of dissolved organic matter and Fe(II) on the abiotic reduction of pentachlorobenzene. Environ Sci Technol 41:7337–7342
Hartzell JL, Jordan TE (2012) Shifts in the relative availability of phosphorus and nitrogen along estuarine salinity gradients. Biogeochemistry 107:489–500. doi:10.1007/s10533-010-9548-9
Heiberg L, Pedersen TV, Jensen HS, Kjaergaard C, Hansen HCB (2010) A comparative study of phosphate sorption in lowland soils under oxic and anoxic conditions. J Environ Qual 39:734–743
Hofmann A, Liang L (2007) Mobilization of colloidal ferrihydrite particles in porous media–An inner-sphere complexation approach. Geochim Cosmochim Acta 71:5847–5861
Hori T, Müller A, Igarashi Y, Conrad R, Friedrich MW (2010) Identification of iron-reducing microorganisms in anoxic rice paddy soil by 13C-acetate probing. ISME J 4:267–278
Howarth R, Paerl HW (2008) Coastal marine eutrophication: control of both nitrogen and phosphorus is necessary. Proc Natl Acad Sci 105:E103
Howarth R, Chan F, Chonley DJ, Garnier J, Doney SC, Marino R, Billen G (2011) Coupled biogeochemistry cycles: eutrophication and hypoxia in temperate estuaries and coastal marine ecosystems. Front Ecol Environ 9:18–26
Hunger S, Benning LG (2007) Greigite: a true intermediate on the polysulfide pathway to pyrite. Geochem Trans 8:1
Jensen HS, Kristensen P, Jeppesen E, Skytthe A (1992) Iron:phosphorus ratio in surface sediments in shallow lakes. Hydrobiology 235(236):731–743
Jiang J, Kappler A (2008) Kinetics of microbial and chemical reduction of humic substances: implications for electron shuttling. Environ Sci Technol 42:3563–3569
Johnston SG, Burton ED, Bush RT, Keene AF, Sullivan LA, Smith D, Martens MA, McElnea AE, Ahern CR, Powell B (2010) Abundance and fractionation of Al, Fe and trace metals following tidal inundation of a tropical acid sulfate soil. Appl Geochem 25:323–335
Kappler A, Straub KL (2005) Geomicrobiological cycling of iron. Rev Mineral Geochem 59:85–108
Kappler A, Benz M, Schink B, Brune A (2004) Electron shuttling via humic acids in microbial iron(III) reduction in a freshwater sediment. FEMS Microbiol Ecol 47:85–92
Keene AF, Johnston SG, Bush RT, Sullivan LA, Burton ED, McElnea AE, Ahern CR, Powell B (2011) Effects of hyper-enriched reactive Fe on sulfidisation in a tidally inundation acid sulfate soil wetland. Biogeochemistry 103:263–279
Khare N, Hesterberg D, Beauchemin S, Wang SL (2004) XANES determination of adsorbed phosphate distribution between ferrihydrite and boehmite in mixtures. Soil Sci Soc Am J 68:460–469
Kirk MF, Roden EE, Crossey LJ, Brealey AJ, Spilde MN (2010) Experimental analysis of arsenic precipitation during microbial sulfate and iron reduction in model aquifer sediment reactors. Geochim Cosmochim Acta 74:2538–2555
Kocar BD, Borch T, Fendorf S (2010) Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite. Geochim Cosmochim Acta 74:980–994
Kopáček J, Hejzlar J, Borovec J, Porcal P, Kotorová I (2000) Phosphorus inactivation by aluminum in the water column and sediments: a process lowering in-lake phosphorus availability in an acidified watershed–lake ecosystem. Limnol Oceanogr 45:212–225
Kopáček J, Borovec J, Hejzlar J, Ulrich K, Norton SA, Amirbahman A (2005) Aluminum control of phosphorus sorption by lake sediments. Environ Sci Technol 39:8784–8789
Lake BA, Coolidge KM, Norton SA, Amirbahman A (2007) Factors contributing to the internal loading of phosphorus from anoxic sediments in six Maine, USA, lakes. Sci Total Environ 373:534–541
Larsen O, Postma D, Jakobsen R (2006) The reactivity of iron oxides towards reductive dissolution with ascorbic acid in a shallow sandy aquifer (Rømø, Demark). Geochim Cosmochim Acta 70:4827–4835
Li X, Cutter GA, Thunell RC, Tappa E, Gilhooly WP III, Lyons TW, Astor Y, Scranton MI (2011) Particulate sulfur species in the water column of the Cariaco Basin. Geochim Cosmochim Acta 75:148–163
Lindsay WL (1979) Iron. In: Lindsay WL (ed) Chemical equilibria in soils. Wiley, New York, pp 1–160
Liu YL, Zhang B, Li CL, Hu F, Velde B (2008) Long-term fertilization influences on clay mineral composition and ammonium adsorption in a rice paddy soil. Soil Sci Soc Am J 72:1580–1590
Lovley DR (1995) Microbial reduction of iron, manganese, and other metals. Adv Agron 54:175–231
Lovley DR (2008) Extracellular electron transfer: wires, capacitors, iron lungs, and more. Geobiology 6:225–231
Lovley DR, Blunt-Harris EL (1999) Role of humic-bound iron as an electron transfer agent in dissimilatory Fe(III) reduction. Appl Environ Microbiol 65:4252–4254
Lovley DR, Phillips EJP, Lonergan J (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments. Environ Sci Technol 25:1062–1067
Lovley DR, Coates JD, Blunt-Harris EL, Phillips EJP, Woodward JC (1996) Humic substances as electron acceptors for microbial respiration. Nature 382:445–448
Lovley DR, Fraga JL, Blunt-Harris EL, Hayes LA, Phillips EJP, Coates JD (1998) Humic substances as a mediator for microbially catalyzed metal reduction. Acta Hydrochim Hydrobiol 26:152–157
Lovley DR, Holmes DE, Nevin KP (2004) Dissimilatory Fe(III) and Mn(IV) reduction. Adv Microb Physiol 49:219–286
Luther GW III, Rickard DT (2005) Metal sulfide cluster complexes and their biogeochemical importance in the environment. J Nanopart Res 7:713–733
Luther GW III, Rozan TF, Taillefert M, Nuzzio DB, Di Meo C, Shank TM, Lutz RA, Craig Cary S (2001) Chemical speciation drives hydrothermal vent ecology. Nature 410:813–816
Luther GW III, Glazer B, Ma S, Trouwborst R, Shultz B, Druschel G, Kraiya C (2003) Iron and sulfur chemistry in a stratified lake: evidence for iron-rich sulfide complexes. Aquat Geochem 9:87–110
Meulepas RW, Jagersma CG, Khadem AF, Buisman CJN, Stams AJM, Lens PNL (2009) Effect of environmental conditions on sulfate reduction with methane as electron donor by an Eckernförde Bay enrichment. Environ Sci Technol 43:6553–6559
Mort HP, Slomp CP, Gustafsson BG, Andersen TJ (2010) Phosphorus recycling and burial in Baltic Sea sediments with contrasting redox conditions. Geochim Cosmochim Acta 74:1350–1362
Nadelhoffer KJ, Emmett BA, Gundersen P et al (1999) Nitrogen deposition makes a minor contribution to carbon sequestration in temperate forests. Nature 398:145–148
Navratil T, Rohovec J, Amirbahman A, Norton SA, Fernandez IJ (2009) Amorphous aluminum hydroxide control on sulfate and phosphate in sediment–solution systems. Water Air Soil Pollut 201:87–98
Nieder R, Benbi DK, Scherer HW (2011) Fixation and defixation of ammonium in soils: a review. Biol Fertil Soils 47:1–14
Norton SA, Coolidge K, Amirbahman A, Bouchard R, Kopáček J, Reinhardt R (2009) Speciation of Al, Fe, and P in recent sediment from three lakes in Maine, USA. Sci Total Environ 404:276–283
Nurmi JT, Trathyek PG (2002) Electrochemical properties of natural organic matter (NOM), fractions of NOM, and model biogeochemical electron shuttles. Environ Sci Technol 36:617–624
Ottley CJ, Davison W, Edmunds WM (1997) Chemical catalysis of nitrate reduction by iron(II). Geochim Cosmochim Acta 61:1819–1828
Pant HK, Reddy KR (2001) Phosphorus sorption characteristics of estuarine sediments under different redox conditions. J Environ Qual 30:1474–1480
Papadas IT, Katerinopoulos L, Gianni A, Zacharias I, Deligiannakis Y (2009) A theoretical and experimental physicochemical study of sulfur species in the anoxic lagoon of Aitoliko-Greece. Chemosphere 74:1011–1017
Pollok K, Hellige K, Harries D, Peiffer S (2009) Redox processes at the nanoscale: a TEM perspective of iron sulphide–iron (oxyhydr)oxide reactions. Geochim Cosmochim Acta 73:A1039–A1039
Poulton SW, Krom MD, Raiswell R (2004) A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim Cosmochim Acta 68:3703–3715
Rakshit S, Uchimiya M, Sposito G (2009) Iron(III) bioreduction in soil in the presence of added humic substances. Soil Sci Soc Am J 73:65–71
Ratering S, Schnell S (2000) Nitrate-dependent iron(II) oxidation in paddy soil. Environ Microbiol 3:100–109
Reitzel K, Hansen J, Andersen F, Hansen K, Jensen H (2005) Lake restoration by dosing aluminum relative to mobile phosphorus in the sediment. Environ Sci Technol 39:4134–4140
Rickard D, Luther GW III (2007) Chemistry of iron sulfides. Chem Rev 107:514–562
Rickard D, Morse JW (2005) Acid volatile sulfide (AVS). Mar Chem 97:141–197
Robinson C, Saunders PW, Madan NJ, Pryce-miller EJ, Pentecost A (2004) Dose nitrogen deposition affect soil microfungal diversity and soil N and P dynamics in a high Arctic ecosystem? Glob Chang Biol 10:1065–1079
Roden EE, Wetzal RG (2002) Kinetics of microbial Fe(III) oxide reduction in fresh water sediments. Limnol Oceanogr 47:198–211
Ruttenberg KC, Sulak DJ (2011) Sorption and desorption of dissolved organic phosphorus onto iron (oxyhydr)oxides in seawater. Geochim Cosmochim Acta 75:4095–4112
Rydin E, Huster B, Welch EB (2000) Amount of phosphorus inactivated by alum treatments in Washington lakes. Limnol Oceanogr 45:226–230
Sahrawat KL (2004a) Organic matter accumulation in submerged soils. Adv Agron 81:169–201
Sahrawat KL (2004b) Ammonium production in submerged soils and sediments: the role of reduction iron. Commun Soil Sci Plant 35:399–411
Sahrawat KL, Narteh LT (2003) A chemical index for predicting ammonium production in submerged rice soils. Commun Soil Sci Plant 34:1013–1021
Schauser I, Chorus I, Lewandowski J (2006) Effects of nitrate on phosphorus release: comparison of two Berlin lakes. Acta Hydrochim Hydrobiol 34:325–332
Scherer HW, Zhang YS (2002) Mechanisms of fixation and release of ammonium in paddy soils after flooding III. Effect of the oxidation state of octahedral Fe on ammonium fixation. J Plant Nutr Soil Sci 165:185–189
Schlesinger WH (2009) On the fate of anthropogenic nitrogen. Proc Natl Acad Sci 106:203–208
Schlesinger WH, Cole JJ, Finzi AC, Holland EA (2011) Introduction to coupled biogeochemical cycles. Front Ecol Environ 9:5–8
Schmidt BHM, Matzner E (2009) Abiotic reaction of nitrite with dissolved organic carbon? Testing the ferrous wheel hypothesis. Biogeochemistry 93:291–296
Scott DT, McKnight DM, Blunt-Harris EL, Kolesar SE, Lovley DR (1998) Quinone moieties act as electron acceptors in the reduction of humic substances by humics-reducing microorganisms. Environ Sci Technol 32:2984–2989
Seitzinger S (2008) Nitrogen cycle out of reach. Nature 452:162–163
Senn DB, Hemond HF (2002) Nitrate controls on iron and arsenic in an urban lake. Science 296:2373–2376
Shenker M, Seitelbach S, Brand S, Haim A, Litaor MI (2005) Redox reactions and phosphorus release in re-flooded soils of an altered wetland. Eur J Soil Sci 56:515–525
Slowey AJ, Brown GE (2007) Transformation of mercury, iron, and sulfur during the reductive dissolution of iron oxyhydroxide by sulfide. Geochim Cosmochim Acta 71:877–894
Smolders AJP, Lucassen ECHET, Bobbink R, Roelofs JGM, Lamers LPM (2010) How nitrate leaching from agricultural lands provokes phosphate eutrophication in groundwater fed wetlands: the sulphur bridge. Biogeochemistry 98:1–7
Steinmann P, Shotyk W (1997) Chemical composition, and redox state of sulfur and iron in complete vertical porewater profiles from two Sphagnum peat bogs. Geochim Cosmochim Acta 61:1143–1163
Straub KL, Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing, nitrate-reducing bacteria from diverse European sediments. Appl Environ Microbiol 64:4846–4856
Straub KL, Benz M, Schink B, Widdel F (1996) Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl Environ Microbiol 62:1458–1460
Straub KL, Schönhuber WA, Buchholz-Cleven BEE, Schink B (2004) Diversity of ferrous iron-oxidizing, nitrate-reducing bacteria and their involvement in oxygen-independent iron cycling. Geomicrobiol J 21:371–378
Struyk Z, Sposito G (2001) Redox properties of standard humic acids. Geoderma 102:329–346
Stumm W, Sulzberger B (1992) The cycling of iron in natural environments: considerations based on laboratory studies of heterogeneous redox processes. Geochim Coamochim Acta 56:3233–3257
Suter D, Banwart S, Stumm W (1991) Dissolution of hydrous iron (III)oxides by reductive mechanisms. Langmuir 7:809–813
Tanwar KS, Petitto SC, Ghose SK, Eng PJ, Trainor TP (2009) Fe(II) adsorption on hematite(0001). Geochim Cosmochim Acta 73:4346–4365
Thorn KA, Mikita MA (2000) Nitrite fixation by humic substances: nitrogen-15 nuclear magnetic resonance evidence for potential intermediates of chemodenitrification. Soil Sci Soc Am J 64:568–582
Weber KA, Urrutia MM, Churchill PF, Kukkadapu RK, Roden EE (2006) Anaerobic redox cycling of iron by freshwater sediment microorganisms. Environ Microbiol 8:100–113
Yin X, Lv X, Jiang M, Zou Y (2010) Research progress of the coupling process of Fe and N in wetland soils. Chin J Environ Sci 31:2254–2259 (in Chinese)
Zak D, Gelbrecht J, Steinberg CEW (2004) Phosphorus retention at the redox interface of peatlands adjacent to surface waters in northeast Germany. Biogeochemistry 70:357–368
Zhang YS, Scherer HW (1999) Ammonium fixation by clay minerals in different layers of two paddy soils after flooding. Biol Fert Soils 29:152–156
Zhang YS, Scherer HW (2000) Mechanisms of fixation and release of ammonium in paddy soils after flooding. II. Effect of transformation of nitrogen forms on ammonium fixation. Biol Fert Soils 31:517–521
Zhang HC, Weber EJ (2009) Elucidating the role of electron shuttles in reductive transformations in anaerobic sediments. Environ Sci Technol 43:1042–1048
Zhang W, Faulkner JW, Giri SK, Geohring LD, Steenhuis TS (2010) Effect of soil reduction on phosphorus sorption of an organic-rich silt loam. Soil Sci Soc Am J 74:240–249
Zhu W, Wang W (2011) Does soil organic matter variation affect the retention of 15NH +4 and 15NO −3 in forest ecosystems? Forest Ecol Manage 261:675–682
Zopfi J, Böttcher ME, Jørgensen BB (2008) Biogeochemistry of sulfur and iron in Thioploca-colonized surface sediments in the upwelling area off central Chile. Geochim Cosmochim Acta 72:827–843
Acknowledgments
The authors thank Prof. Max Haggblom at Rutgers, the State University of New Jersey, and anonymous reviewers for valuable comments. This work was financially supported by the grants from the “Knowledge Innovation Programs” (KZCX2-YW-JC402) and the “Hundred Talent Program” (no. A0815) of the Chinese Academy of Sciences, the Ministry of Science and Technology of China (2009DFB90120), and the National Science Foundation of China (no. 40871244).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Thomas DeLuca
Rights and permissions
About this article
Cite this article
Li, Y., Yu, S., Strong, J. et al. Are the biogeochemical cycles of carbon, nitrogen, sulfur, and phosphorus driven by the “FeIII–FeII redox wheel” in dynamic redox environments?. J Soils Sediments 12, 683–693 (2012). https://doi.org/10.1007/s11368-012-0507-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-012-0507-z