
Abstract
Purpose
The current study was conducted in order to assess the release of dissolved contaminants from sediment relocation works in Oslo harbour, Norway, whilst operations were being carried out, and to assess the potential for spreading into the wider fjord area.
Materials and methods
This was achieved by comparing the concentration of metals and organic contaminants in the vicinity of the disposal site, and at strategic locations in the adjacent fjord area. In total, 14 stations were chosen, with passive sampling devices, diffusive gradients in thin films for metals and semipermeable membrane devices for organic compounds, deployed at various depths at each station for a period of 1 month.
Results and discussion
The highest concentrations were generally found closer to the harbour area where trace metals and organic contaminants are supplied from several sources including river discharges and old, contaminated sediments. Near the deposition site, the concentrations of polychlorinated biphenyls were low and the concentration gradients for Cd, Zn and Cu indicated removal of these metals from the water column, probably due to association with sulphides present at the deposition site or in the discharged sediments. Elevated concentrations of Pb and polycyclic aromatic hydrocarbons (PAHs) revealed that the disposal acted as a source of these compounds, but maximum concentrations were similar to or less than those predicted before the deposition begun. The estimated vertical transport and spreading to areas outside of the semi-enclosed sill basin was negligible.
Conclusions
Strictly, the validity of the results presented here are limited to a 1-month period of the disposal operations which lasted nearly 3 years. During this period, release of contaminants was compound-specific, but even for the most labile PAHs and Pb, the release appeared to be in the same order of magnitude or smaller than the release from other local sources not limited in time to the duration of the remediation operation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Marine sediments primarily act as a sink for particle-bound contaminants. Therefore, harbour sediments have become worldwide hotspots of contaminants from shipping activities and run-off from urban and industrialised areas (Förstner and Salomons 1988; Bortone et al. 2004). During regular harbour management, bottom impacts and re-suspension from propeller activity are frequent occurrences which make the sediments a secondary source with significant potential for desorption and spreading of the most bioavailable, dissolved contaminant fractions (Hedge et al. 2009). Dredging is a universal activity motivated by construction works, maintenance of shipping depths or environmental objectives related to waterfront renovation and long-term reduction of contaminant transfer to biota. However, during dredging operations, the risk of contaminant spreading increases on a short-term basis not only due to dredging itself, but also due to subsequent handling, transportation and disposal (Thibodeaux et al. 2004; Wenning et al. 2006; Alvarez-Guerra et al. 2008).
In the Oslo harbour remediation project, Norway, 440,000 m3 of contaminated sediments were removed by grab dredging during the period February 2006 to October 2008. The contaminated sediments were transported in barges to a sub-sea deposit at 70 m depth in a semi-enclosed basin located adjacent to the harbour area. Here, the barges were unloaded by pumping through a closed pipe and discharged via an energy dissipater attached to the open end of the pipe a few meters above the bottom, in a naturally confined depression in the southern end of the basin. Subsequently, the deposition area is being covered with a 40-cm-thick layer of clean sand to reduce leakage and contaminant uptake in biota.
During discharge, dissolved, desorbed and particle-bound contaminants may spread from recently deposited material and particles suspended in the water surrounding the discharge point. Thus, as a supplement to the general monitoring programme for the remediation operation (Pettersen and Breedveld 2009), this paper presents results from a separate investigation on dissolved contaminants performed during a short period about midway in the operation. Dredging and deposition activities were carried out as normal during the sampling period, i.e. daily deposition of one to three barge loads with up to 400 m3 of dredged sediment in each barge (Løken et al. 2007).
Organic contaminants and metals were collected on passive sampling devices (PSDs) deployed strategically near the discharge site and at selected stations near to the harbour and city centre, and at reference sites assumed not to be influenced by known point sources. The primary benefit of using PSDs is their ability to provide time-integrated information about fluctuating water concentrations (Gale 1998; Gourlay-France et al. 2008; Hawker 2010). This is of special relevance here as concentrations may vary with the level of depositional activity. Additionally, they often allow lower detection limits (Lebo et al. 1995; Harman et al. 2008) and sample only the biologically relevant freely dissolved fraction. The semipermeable membrane device (SPMD) (Huckins et al. 1990) was chosen to measure organic contaminants as these have been extensively used in monitoring studies (Esteve-Turrillas et al. 2008), and models explaining chemical uptake are available (Huckins et al. 2006). A further advantage of SPMDs is that concentrations can be corrected for environmental factors such as water turbulence and temperature, which influence uptake by the use of performance reference compounds (PRCs) (Booij et al. 1998; Huckins et al. 2002). For metals, the diffusive gradient in thin films (DGTs) sampler was used (Davison and Zhang 1994; Zhang and Davison 1995). These samplers bind metals in an ion exchange sorbent packed behind a filter and a diffusion gel and have been successfully applied to a wide range of environmental monitoring scenarios (Warnken et al. 2007).
Thus, the dual objective of the current study was to measure the freely available fraction of metals and hydrophobic organic compounds using a comprehensive passive sampling approach, and to use the data to examine the extent to which the contaminants were spreading from the disposal site in comparison with other potential sources in the harbour area.
2 Materials and methods
2.1 Study location
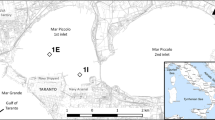
The Bekkelagsbassenget (Fig. 1) is a semi-enclosed basin surrounded by islands and sounds with sill depths of 31–42 m. Figure 2a presents typical temperature and salinity profiles for the summer season, where the strongest stratification is found in the upper 20 m, with a more homogeneous water mass below. In such enclosed basins, tidal movements create only weak currents below sill depth. During a period of 1 month, Skei et al. (1999) reported a mean current speed of 0.5 cm s−1 at 67 m depth close to station BN1. Higher current speeds (above 1 cm s−1) occur occasionally during inflows of heavier water and deep water renewal. Historically, oxygen conditions have been poor in the deep water, and over the years, the basin has been used for dumping of various anthropogenic wastes. In recent years, oxygen conditions have improved (see Fig. 2b) partly as result of a relocation of a 1 m3 s−1 discharge of treated sewage water to a depth of 50 m in the northeast of the basin (Berge et al. 2010). The 0.3-km2 area that is regulated for deposition is a natural depression in the southern part of the basin. It is separated from the surrounding basin by a low underwater ridge or sill which has a maximum depth of approximately 65 m. Comparison of maximum concentrations of Hg and polychlorinated biphenyls (PCBs) observed in the surface sediments in Bekkelagsbassenget in 1998 (Skei et al. 1999) and December 2007 (Skei and Nilsson 2008) indicated a small increase from 0.7 to 1.0 μg Hg g−1 and 0.024 to 0.044 μg g−1 ∑PCB7 (“seven Dutch”).

Map and schematic bottom profile showing stations and sampling depths for passive samplers deployed in the inner Oslofjord in May–June 2007. The shaded areas show the deposition site in Bekkelagsbassenget and the remediation area in the inner part of the harbour basin within which both dredging and capping has been undertaken

Temperature, salinity, oxygen and turbidity profiles measured on the 29th of May 2007 in Bekkelagsbassenget. (Data from Magnusson et al. 2008)
2.2 Sampling
Passive samplers were mounted on rigs deployed at 14 stations in the Inner Oslofjord from the 10th of May 2007 to the 12th of June 2007 (Fig. 1, Table 1). Each rig consisted of a double mooring at the bottom and a line tied to floats about 5 m above the top sampler. Samplers were mounted in stainless steel containers (EST labs, St Joseph, MO, USA) at fixed distances (3, 10, 20 and 30 m) from the mooring, except for one reference station (RS15) at which depths were chosen to correspond to 3 and 10 m above the depth of the closest basin sill. In order to maximise resources and increase the coverage of the measurements, it was decided to sample at several depths, but with only a single sampler of each type deployed at each depth. Relative standard deviations between sampler replicates is typically about 10% and less than both spot sampling and biological methods (Harman et al. 2011). Additionally, the PRC results from individual samplers confirm that SPMDs were not damaged.
Two rigs, BN0 and SW4 were not found during retrieval. Because of the loss of BN0, probably during relocation of disposal platform or moorings, BN1 became the rig located closest to the deposition site at the relevant depth (see Fig. 1). Particle spreading was frequently observed beyond the north-eastern boundary of the deposit site (Pettersen and Breedveld 2009). Data recorded by Magnusson et al. (2008) during our PSD deployment (see Fig. 2b), confirmed elevated turbidity in the deep water at both BN0 and BN1 so that samples 14 (BN1-58) and 15 (BN1-65), and possibly also samples 13 (BN1-48) and 11 (BN4-52), had been exposed to particles from the deposit site.
The remaining 12 rigs were located within Bekkelagsbassenget (BN1, BN4, BN8), at basin sills (SW8, SS1, SE1), in the shallow harbour basin (HN10, HN15, HN20) and in reference areas south and west of the operation area (RS15, RW20, RW30). In addition to the deep water discharge, irregular spills in the surface layer at the deposit site and dredging activity near the mouth of the river Akerselva were considered to be critical points at which contaminants may have been spread during our investigation. The sediments throughout the harbour basin are generally contaminated with most of the compounds analysed in this investigation (Schaanning et al. 2005). Therefore, recycling from non-remediated sediments may be an important contaminant source in the area as well as regular harbour activities, urban runoff, river discharge and a sewage water treatment plant discharge near BN8. An important objective of the deposition operation was to avoid spreading of contaminants from the deposit site to the fjord outside Bekkelagsbassenget. The samplers positioned inside the basin and in the sill areas were primarily intended to reveal any such contaminant transport.
Before the disposal operation begun, a test discharge of 400 m3 of contaminated sediments was performed on the 29th of November 2005 by opening a barge loaded with dredged harbour sediments in the surface water in the deposit area. Samples were collected from depths of 5–68 m before the discharge and during a period of 0.5–6 h after the discharge. The samples had particle loads ranging 0.3–85 g L−1 and the analyses of filtered and unfiltered samples before and after the test discharge revealed a significant potential for contamination of the water column (Table 2). A complete description of that survey and analyses is reported in Schaanning et al. (2006b).
2.3 Chemical analysis
DGTs for metals sampling were supplied from DGT Research LTD (Lancaster, UK). The samplers consisted of a polyacrylamide hydrogel with ion exchange Chelex resin. After removal of the gel, the resin was eluted in 1 mL conc. HNO3 and diluted ten times with ultrapure water (Garmo et al. 2003). The trace metals were analysed by inductively coupled plasma-mass spectrometry using a PerkinElmer Sciex ELAN 6000.
SPMDs spiked with deuterated PRCs (acenaphthene-d10, fluorene-d10, phenanthrene-d10 and Chrysene-d12) were delivered from ExposMeter (Travelsjo, Sweden). After retrieval, the SPMD surface was carefully rinsed before dialysis with 2 × 150 mL hexane. The extracts were reduced with nitrogen and cleaned using gel permeation chromatography before being divided into two fractions for analysis of polycyclic aromatic hydrocarbons (PAHs; including the PRCs) and PCBs. The PCB fraction received further clean up by partitioning with concentrated sulphuric acid. The PAH fraction was analysed by gas chromatography–mass spectrometry and the PCB fraction by gas chromatography with electron capture detection. An Agilent Technologies 6890GC (Santa Clara, USA) equipped with a 30-m column for PAHs or a 60-m column for PCBs. Both columns had a stationary phase of 5% phenyl methylpolysiloxane (0.25 mm internal diameter and 0.25 μm film thickness, Agilent Technologies). Quantification of individual components was conducted by the relative response of internal standards. Further descriptions of procedures for extraction, clean-up and chemical analysis can be found in Harman et al. (2008).
2.4 Quality assurance
Field control SPMDs were exposed to air to check for contamination during deployment and retrieval operations. Laboratory control SPMDs followed exposure from solvents, glass equipment, laboratory procedures, etc. and at least one of each type of control was used per ten exposed samplers. Initial concentrations of PRCs were determined as the mean concentration in laboratory control SPMDs. With the exception of small amounts of naphthalene (ca. 40 ng per SPMD) and gamma-hexachlorocyclohexane (γHCH, 3 ng per SPMD), no target compounds were found in the control SPMDs. The presence of naphthalene in unexposed SPMDs has been reported by others (Boehm et al. 2005; Harman et al. 2009). The fjord samples had quantifiable levels of naphthalene at two sampling stations only, but because of frequent occurrences of γHCH at similar levels as those found in the laboratory controls, the results for this compound are not considered further. No other contaminants were quantifiable in any control samples, indicating that SPMDs had not been exposed to target compounds during deployment retrieval, transport or storage.
2.5 Calculation of water concentrations
For the metals, the accumulated mass M is measured after a known deployment time t, and then Eq. (1) can be used to calculate the water concentration C:
where D is the diffusion coefficient, A is the surface area of the sampler and Δg is the thickness of the diffusion layer (Zhang and Davison 1995). Further details concerning the determination of diffusion coefficients and correction for temperature are given by Zhang and Davison (1995).
Accumulation of hydrophobic organic contaminants by SPMDs can be explained by an initial linear and integrative uptake phase followed by curvilinear and equilibrium partitioning stages. A model describing uptake at any stage was used to calculate freely dissolved water concentrations (C w) from SPMD data (Huckins et al. 1993):
where N is the absorbed amount (nanograms), V s is the volume of the sampler (cubic centimeters), K sw is the SPMD water partitioning coefficient (cubic centimeter per cubic centimeter) and R s is the apparent water sampling rate (liters per day). Log K sw were estimated from the log K ow of PRCs (and subsequently, target compounds) using the quadratic equation provided by Huckins et al. (2006):
where the intercept a 0 = −2.61, for most hydrophobic compounds. Sampling rates were determined in situ, by the use of PRCs and an empirical model, described in detail by Huckins et al. (2006). PRCs where the remaining amount after deployment was between 10–90% of starting concentrations were used in the model. The generated R s values were then used in Eq. 2 to calculate water concentrations for target compounds, with K sw derived from Eq. (3). A more detailed description of how we apply the PRC data is given elsewhere (Harman et al. 2009).
2.6 Statistics
Concentrations of metals and organic contaminants from the different areas were compared statistically using analysis of variance (ANOVA), grouped by region. Where ANOVA revealed significant differences, groups were compared using Tukey’s post-hoc test comparing all groups or Dunnett’s test comparing with reference group. The level of significance for rejection of H0 “no difference between groups” was set to 0.05.
Principle components analysis (PCA) for the relative contribution of each compound was calculated separately for the three groups of compounds: metals, PAHs and PCBs. PCA plots are frequently used as a forensic tool in environmental work by revealing source-characteristic component patterns and transportation pathways (Zitko 1994; An et al. 2010). If all compounds are conservative, i.e. concentrations are affected by no other processes than water mixing, then PCA plots may be used as a hydrographic tool to identify water mixing.
Except for the concentrations used for calculating principal components for PCBs, concentrations below the limits of detection (<D.L.), were set to zero and stations without detectable components were excluded from the analysis. The statistical software JMP® 9.0.0 from SAS Institute Inc. was used for all analyses presented in the present study.
3 Results
3.1 Metals
The highest concentrations of Zn and Cd occurred in sample 1 (HN20-15) collected in the north-eastern part of the harbour area, and the lowest occurred in samples 15 (BN1-65) and 14 (BN1-58) collected near the discharge of the dredged sediments (Fig. 3). For Cu, Pb and Ni, high concentrations were determined in sample 5 (HN10-27) collected in the southern part of the harbour basin and in at least one of the samples 11, 14 or 15, which were all collected in the Bekkelagsbassenget below 50 m depth and potentially influenced by the discharge (see Fig. 3). Notably, sample 15 (BN1-65), which was located nearest to the discharge point where the highest turbidities were observed (see Fig. 2), showed low concentrations of all metals.

Statistical comparison (ANOVA, Tukey comparison) of the metal concentrations in five groups of samples: near deposit site (BN1), Bekkelagsbassenget (BN4 and BN8), sill stations (SE1, SS1, SW4), harbour basin (HN10, HN15, HN20) and reference (RS15, RW20, RW30) showed that the concentration of Pb in the harbour was significantly higher (p < 0.05) than that found at both the reference and the Bekkelagsbassenget stations. For the other metals analysed (Cd, Cu, Ni and Zn), there were no significant difference (p > 0.05) between these groups of stations.
In the PCA (Gabriel 1971) for the relative concentration of the five metals determined, the two first components explained 71.2% of the total variance (Fig. 4). The biplot shows that Cu, Ni and Zn contributed primarily in component 1, whereas Cd and Pb contributed primarily in component 2. The station plot shows most of the samples grouped in an elliptical cluster between the two reference samples 22 and 24. Four of the samples located outside the main cluster (11, 13, 14 and 15) were collected in the vicinity of the discharge point. Samples 11 and 14 occurred near a straight line connecting samples 13 and 15. This would be the case if samples 11 and 14 represent a mixture of samples 13 and 15 and provided that mixing was the main process affecting the metal concentration. Figure 1 shows that the spatial relationships are suitable to support this interpretation. Sample 5, however, was collected in more shallow waters north of the basin and the deviating component composition was more likely a result of local sources or processes, rather than any impact from discharge or dredging operations.

Principal component analyses of relative concentrations of 5 metals (top), 19 PAH compounds (middle) and 10 PCBs (bottom). Stations are represented by points in the left-hand diagrams. The length and direction of arrows in the right-hand diagrams indicate the loadings from each compound. For PCBs, the detection limits (0.01–0.07 ng L−1) were used for concentration of components not detected; note PCB 105 is masked by the group of PCBs 153, 209, 101 and 156, to the right. PAH abbreviations: NAP naphthalene, ACY acenaphthylene, ACE acenaphthene, FLE fluorene, DBT dibenzothiophene, ANC anthracene, PHA phenanthrene, FLA fluoranthrene, PYR pyrene, BAA Benz[a]anthracene, CHR chrysene, BBF benzo[b]fluoranthrene, BFK benzo[k]fluoranthrene, BEP benzo[e]pyrene, BAP benzo[a]pyrene, PER perylene, BGP benzo[ghi]perylene, DBA dibenz[a,h]anthracene, IDP indeno[1,2,3-cd]pyrene
The non-conservative behaviour yielding the “extreme” metal compositions in samples 13 and 15 occurred near the discharge of harbour sediments and within the part of the water column in which elevated turbidity was observed (see Fig. 2). The discharge location is often oxygen-deficient and water sampled <5 m above the bottom during the test discharge in 2005 appeared anoxic. Sulphides, which may also have been abundant in the discharged harbour sediment, react with many metals to form almost insoluble metal sulphides (Du Laing et al. 2009). If sulphide was present in the sediments, bottom water or discharged material, the observed concentration gradients of Cd, Zn and Cu at station BN1 were as expected. One problem with this interpretation is that the concentration of Pb, which forms similarly insoluble metal sulphides, increased in the same area. In estuaries, high concentrations of dissolved Pb have been associated with increased turbidity (Martino et al. 2002), and in oxic environments, desorption from re-suspended sediments is known to provide a general increase of dissolved concentration of both metals and organic ligands (Turner et al. 1998; Millward and Liu 2003). Cobela-Garcia and Prego (2004) attributed different behaviour between Pb and Zn to the presence of a high-stability Pb-organic complex, but it is not clear how such complexes may interfere with sulphide precipitation and uptake on the DGT sampler.
In the upper part of the layer affected by re-suspended sediments (sample 13), sulphide will be less abundant and dissolved metals may escape sulphide precipitation. The data in Table 2 showed that during the test discharge in the deposition area, the concentration of Pb in filtered samples was only 2% of the total concentration compared to a dissolved fraction of 27–29% for Cu and Zn and 59% for Cd. These samples were collected throughout the mainly oxic water column and fit well with the DGT observations in sample 13.
3.2 Polycyclic aromatic hydrocarbons
The concentration of the sum of the 16 most commonly measured PAH components (EPA16) varied between 2.3 and 13.6 ng L−1 ∑PAH16 (see Table 2). This was more than the background concentration determined in discrete water samples before the test discharge, but less than the concentrations determined after the test discharge. The SPMD concentrations compared well with other investigations performed during the disposal operation. Thus, polyoxymethylene (POM) equilibrium passive samplers deployed at sill depth near the discharge in April 2007 showed 14–29 ng L−1 ∑PAH16 (Oen and Hauge 2007; Cornelissen et al. 2008b), and SPMDs deployed by Bergqvist and Zaliauskiene (2007a), showed typical ∑PAH16 concentrations of about 10 ng L−1 (range 5–40 ng L−1) for stations in Bekkelagsbassenget and 5 ng L−1 at a reference station south of the basin. Compared with national criteria for fjord and coastal water, the concentrations of PAH determined by SPMDs corresponded to between 1/5 (pyrene) and 1/1,000 (acenaphthene) of the upper limit for class II “no toxic effects” (SFT 2007).
The highest concentrations of ΣPAH19 were observed in samples 3 and 4 near the outlet of the Alna River and in samples 13–15 collected close to the discharge point. Bergqvist and Zaliauskiene (2007a) observed a higher concentration of 140 ng L−1 ΣPAH16 in a sampler deployed in shallow water in Bjørvika near the mouth of Akerselva, the other major river discharging in the harbour area. Both rivers are known to be significant sources of PAHs and PCBs (Ranneklev et al. 2009). The most common congeners were pyrene, which contributed an average of 39% of the 19 PAHs determined, followed by 16% for fluoranthene and 10% for phenanthrene. Acenaphthene contributed on average 9%, but in sample 14 collected near the discharge point this congener contributed as much as 23% of the total ΣPAH19 (see Fig. 3). Phenanthrene showed a distribution pattern similar to acenaphthene with relative low concentrations in the harbour area and higher concentrations at the deposition site. Because of the altered congener composition, PCA may be favourably used for identification of PAHs spreading from the discharge.
Statistical comparison (ANOVA, Dunnett’s test) of the same five groups of samples as used for comparison of metal concentrations showed that compared to the reference (all R-samples), the concentration of ∑PAH19 were significantly (p < 0.05) higher both in the harbour (all H-samples) and at the deposit site (all BN1-samples). Interestingly, however, the lighter weight compounds with octanol–water partition coefficients (log K ow) ranging from 3.3 to 4.2 (naphthalene, acenaphthene, fluorene and dibenzothiophene) were significantly higher (p < 0.05) only at the deposit site (all BN1-samples), whereas the heavier compounds with log K ow ranging from 5.1 to 6.8 (fluoranthene, pyrene, benz[a]anthracene, chrysene, benzo[b,j]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, perylene, indeno[1,2,3-cd]pyrene and dibenzo[ac/ah]anthracene) were significantly higher only in the harbour area (all H-samples). This confirmed that re-suspension of old harbour sediments gives a shift in water concentrations towards more water-soluble PAHs. In the other basin samples (BN4 and BN8) and sill area (S), neither ∑PAH19 nor any individual PAH compound was significantly different from the reference samples.
The plot of the principal components, showed that most of the samples occurred in a cluster between reference samples (22, 23 and 24) upwards to the left and the two samples 3 and 4 collected in the harbour area near the outlet of river Alna downwards to the right (see Fig. 4, middle left). Similar to the PCA plot for the metals, the two samples influenced by suspended harbour sediments (13 and 15) also occurred at “extreme” positions outside the main cluster (up, right and down, left), with samples 11 and 14 again positioned near the 13–15 straight line indicating that the these two samples obtain their PAH composition primarily from mixing of the water sampled at 13 and 15. The biplot arrows showed that samples 3 and 4 occurred at a position characterised by relatively high contributions from four-ring compounds fluoranthene (%FLA), chrysene (%CHR) and benz[a]anthracene (%BAA). These compounds have previously been used to discriminate between near-source and diffusely contaminated sediments in the Oslofjord region (Næs and Oug 1997).
Sample 15 had a PAH composition characterised by relatively high abundances of five-and six-ring PAHs compounds (benzo[ghi]perylene, benzo[e]pyrene, dibenz[ac/ah]anthracene, indeno[1,2,3-cd]pyrene and perylene), as opposed to sample 13 which had typically higher fractions of the more water-soluble three-ring PAHs (acenaphthene, anthracene and dibenzothiophene). Thus, both the metals and PAHs showed that components with low affinity towards particles and organic complexes (Cd, three-ring PAHs) were more abundant at 48 m depth, in the upper the part of the water column influenced by discharged harbour sediments, than near the bottom where the least water-soluble components (Pb and five- and six-ring PAHs) had accumulated.
3.3 Polychlorinated biphenyls
PCBs were frequently <D.L. of 0.01–0.07 ng L−1. Thus, PCB101, 105, 156 and 209 were <D.L. in all samples and all PCB congeners analysed were <D.L. in 12 of the 24 samples collected. PCB52 was one of the most frequently detectable congeners with up to 0.04 ng L−1 in samples collected in the harbour basin. The highest concentration of ∑PCB7 was 0.203 ng L−1. This was consistent with the observation of dissolved concentration <0.2 ng ∑PCB7 L−1 obtained from filtered water samples collected after the test discharge (see Table 2). Furthermore, the current PCB data agreed well with the concentrations determined using POM samplers (see previous section) deployed in the same area a few months prior to our investigation (Oen and Hauge 2007; Cornelissen et al. 2008b) and the concentrations of 0.02–0.05 ng PCB52 L−1 determined from SPMDs deployed near the discharge in 2006 (Bergqvist and Zaliauskiene 2007b).
Figure 3 shows that ΣPCBs were detectable in all samples from the harbour area. Maximum concentrations occurred in samples 2 and 4 collected 3 m above the bottom in either end of the harbour area and were nearly twice as high as the concentrations in samples 1 and 3 collected 10 m above the bottom at the same stations. Outside of the harbour area, the highest concentration was 0.05 ng L−1 in sample 15 collected close to the discharge site.
In the construction of the PCA plot in Fig. 4, detection limits were used for non-detectable concentrations. The figure shows that four samples from the harbour area (1–4) were displaced from the bulk of samples clustered near the origin. The biplot arrows showed that this displacement resulted primarily from contributions from low molecular weight compounds PCB28, 52, 118 and 138.
4 Discussion
4.1 Contaminant sources in the harbour area
The Akerselva and Alna Rivers are major sources for the input of organic contaminants to the inner Oslofjord (Ranneklev et al. 2009). Sediment data does, however, show decreasing trends for trace metals (Lepland et al. 2010) and PCBs (Arp et al. 2011). Decreased input has been confirmed by Berge et al. (2008) who showed that during the period 1992–2008, the concentration of PCBs within the top layer of the sediments in the inner Oslofjord had decreased by an average factor of 8.6 at 22 stations. The data from stations HN15 and HN20 confirm, however, that river discharge and recycling from contaminated harbour sediments remain two important sources contributing to increased dissolved concentrations in the fjord water.
In a mass balance budget for contaminant fluxes in Oslo harbour, Schaanning et al. (2005) estimated an annual sedimentation in the eastern part of the harbour, near station HN15, of 0.14 mg PCB m−2 year−1and 29 mg PAH m−2 year−1. River discharges were found to account for 60–80% of this sedimentation. In the western part of the harbour area, near HN20, sedimentation rates were smaller (0.06 mg PCB m−2 year−1and 5 mg PAH m−2 year−1) and river discharge could only account for 10–20%. Sediment concentrations were, however, not much different at the two stations (HN15 and HN20) and calculated fluxes from the sediments were of similar magnitude (Schaanning et al. 2005). The significance of the sediment flux was confirmed for PCBs which showed similar concentrations at the two stations and higher concentrations at 3 m compared to 10 m above the bottom (see Fig. 3). For PAHs, however, the concentrations were clearly lower at station HN20 located in the western harbour basin, less influenced by the river discharges, and in particular in the sample closest to the bottom. It appears that the river discharge is an important source of dissolved PAHs in the harbour basin water, whereas the contaminated bottom sediment is a more important source for PCBs.
The flux from these sediments are in the order of 1 μg m−2 year−1 dissolved PCBs (Schaanning et al. 2006a, b; Eek et al. 2008, 2010), which is small compared to local river discharge and sedimentation rates in the order of 100 μg m−2 year−1 total PCBs (Schaanning et al. 2005). The strong sediment signal for PCBs found in samples near the sediment surface in the harbour area seems to suggest that the flux of dissolved PCBs from sediments contribute more to the ecological risk than the much higher total flux from the rivers. It also appeared to suggest that un-remediated sediments in the outer harbour basin may be an important source for PCBs to organisms living in the fjord waters. These sediments are not easily available for dredging, but capping has been considered appropriate, and applied in some areas.
4.2 Release from deep discharge of dredged sediments
In October 2006, shortly after the deposition had begun, increased turbidity and concentrations of dissolved PCBs and PAHs were found 5 m above the bottom at several stations in Bekkelagsbassenget and extending upwards in the water column to 53 m depth near the discharge (Cornelissen et al. 2008a, b). In the present study, any further spreading of contaminants released from the discharged sediments would be expected to yield elevated concentrations first at BN1 (samples 12–15), then at the more remote basin stations BN4 and BN8 (samples 6–11) and ultimately at the sills (samples 16–18) and in case of northwards transport, even sample 5 at 27 m depth at HN10 might perhaps be affected.
The PCA plots both for metals and PAHs revealed some similarity between sample 5 and the near-discharge sample 15, and the occurrence of sample 5 close to the straight line between sample 15 and samples 3 and 4 might suggest influence from both river discharge and sediment disposal. However, the large difference in depth and horizontal distance between the two samples, the lack of consistency between the PAH and metal-PCA plot, and the lack of any signals from other basin samples collected at intermediate locations (stations BN4, BN8) makes the evidence for northwards migration of contaminants beyond BN4 (sample 11) very weak. In contrast to sample 5, the discharge impact on sample 11 was consistent both with the spatial position (neighbouring station, same depth), elevated concentrations of PCBs, PAHs, Cu and Ni, and that the PCA plot for metals indicated vertical mixing of water sampled near the discharge location (samples 13, 14 and 15). It can also be added that a northward transport of contaminants to the outer fjord via the harbour basin is not likely due to the topographical restrictions posed by shallow sills at about 25 m depth and narrow passage to the outer fjord west of HN20. This leaves southward transport through the sounds as the only possible pathway for significant spreading of contaminants from Bekkelagsbassenget to the outer fjord.
The PSDs in the sill areas (samples 16–18) gave no evidence for any transport of contaminants from the disposal site to the outer fjord, neither by elevated concentrations (see Fig. 3) nor by deviations in the component patterns (see Fig. 4). This was consistent with the vertical distribution of contaminants inside the Bekkelagsbassenget, which showed that concentration gradients of Pb and PAHs extended upwards to ≥45 m, which was below the basin sill depths of ≤42 m. Nevertheless, the vertical distribution indicated some upwards transport of dissolved contaminants, and the profile of benzo[a]pyrene (BAP) (see Fig. 3) was used for estimation of this transport. Assuming diffusion coefficients of 0.06–0.12 cm2 s−1 the observed concentration gradient gave vertical transport of 2.6–5.2 μg BAP m−2 year−1. If this flux was maintained throughout the deposition period (32 months) and across the entire basin area at sill depth (3.53 km2), the total transport to sill depth would be 25–50 g. In a model prepared for the environmental impact assessment for the deep water disposal, a loss of 30 g benzo[a]pyrene by diffusion to sill depth and lateral transport across the sills was predicted (Schaanning and Bjerkeng 2001; Bjerkeng et al. 2002). The predicted concentrations in the basin water near the discharge were in the same order of magnitude as those observed here for benzo[a]pyrene (0.1 ng L−1) and PCB (0.01 ng L−1). For the other contaminants included in the model, i.e. dissolved pyrene, Cd, Zn, Cu and Pb, concentrations were four to 50 times higher than those observed in the present study (Bjerkeng et al. 2002).
The PCA plots revealed non-conservative behaviour of both PAHs and metals within the water mass contaminated with particles from the disposal site with more soluble components such as Cd and three-ring PAHs dominating in the upper part of the contaminated layer, and components with higher affinity to particles and organic complexes such as Pb and four- and five-ring PAHs dominating near the bottom. In addition, removal by metal sulphide precipitation could explain low concentrations of dissolved Cd, Zn and Cu near the bottom.
5 Conclusions
For source identification and water quality assessment, PSDs combine the precision of the chemical analyses of discrete water samples and the time integration of exposure provided by biomonitoring organisms. The PSDs proved suitable to identify important sources and sinks for dissolved, bioavailable contaminants in the inner Oslofjord. The sediment discharge near the bottom at the disposal site was a clear source of dissolved Pb, PAHs and some PCBs, but a sink for dissolved Cd and Zn. However, even for the most labile compounds, estimated concentrations in the bottom water and spreading via upwards diffusion and lateral transport across the basin sills was similar to or smaller than the concentrations and spreading predicted before the remediation operation begun. Relatively high concentrations measured in the harbour basin indicated that river discharge and release from sediments outside the remediated area were important sources for dissolved PAHs and PCBs. These sources appeared large compared to the deep water discharge and will remain active long after the termination of the remediation operation.
References
Alvarez-Guerra M, Alvarez-Guerra E, Alonso-Santurde R, Andres A, Coz A, Soto J, Gomez-Arozamena J, Viguri JR (2008) Sustainable management options and beneficial uses for contaminated sediments and dredged material. Fresenius Environ Bull 17:1539–1553
An Q, Wu Y, Wang Z, Li Z (2010) Assessment of dissolved heavy metal in the Yangtze River estuary and 1st adjacent sea, China. Environ Monit Assess 164:173–187
Arp HPH, Villers F, Lepland A, Kalaitzidis S, Christanis K, Oen AMP, Breedveld GD, Cornelissen G (2011) Influence of historical industrial epochs on pore water and partitioning profiles of polycyclic aromatic hydrocarbons and polychlorinated biphenyls in Oslo harbour, Norway, sediment cores. Environ Toxicol Chem 30:843–851
Berge JA, Magnusson J, Schøyen M, Skei J (2008) Investigations of contaminants in sediments from Bunnefjorden. Norwegian Institute for Water Research, Report 5583–10, ISBN 978-82-577-5318-4, p 58 (www.niva.no)
Berge JA, Amundsen R, Bjerkeng B, Bjerklnes E, Espeland SH, Gitmark JK, Holth TF, Hylland K, Imrik C, Johnsen T, Lømsland ER, Magnusson J, Nilsson H, Rohrlack T, Sørensen K, Walday M (2010) Monitoring the pollution status of the inner Oslofjord 2009. Norwegian Institute for Water Research, Report 5985–10, ISBN 978-82-577 -720-5, p 145 (www.niva.no)
Bergqvist PA, Zaliauskiene A (2007a) Investigation of chemicals released from Malmøykalven dumping area. Polycyclic aromatic hydrocarbons. ExposMeter AB, Tavelsjö, Sweden. Report nr. 2006 N-003, p 59
Bergqvist PA, Zaliauskiene A (2007b) Investigation of chemicals released from Malmøykalven dumping area. Polychlorinated biphenyls. ExposMeter AB, Tavelsjö, Sweden. Report nr. 2006 N-001, p 46
Bjerkeng B, Schaanning M, Tobiesen A (2002) Opprydding av forurensede sedimenter - Risiko for skadelige effekter på organismer under etablering av dypvannsdeponi Malmøykalven. NIVA-Technical MEMO, 05.11.2002, p 13
Boehm PD, Page DS, Brown JS, Neff JM, Bence AE (2005) Comparison of mussels and semi-permeable membrane devices as intertidal monitors of polycyclic aromatic hydrocarbons at oil spill sites. Mar Pollut Bull 50:740–750
Booij K, Sleiderink H, Smedes F (1998) Calibrating the uptake kinetics of semipermeable membrane devices using exposure standards. Environ Toxicol Chem 17:1236–1245
Bortone G, Arevalo E, Deibel I, Detzner H-D, de Propris L, Elskens F, Giordano A, Hakstege P, Hamer K, Harmsen J, Hauge A, Palumbo L, van Veen J (2004) Sediment and dredged material treatment—synthesis of the SedNet work package 4 outcomes. J Soils Sediments 4:225–232
Cobela-Garcia A, Prego R (2004) Chemical speciation of dissolved copper, lead and zinc in a ria coastal system: the role of resuspended sediments. Anal Chim Acta 524:109–114
Cornelissen G, Pettersen A, Broman D, Mayer P, Breedveld GD (2008a) Field testing of equilibrium passive samplers to determine freely dissolved native polycyclic aromatic hydrocarbon concentrations. Environ Toxicol Chem 27:499–508
Cornelissen G, Arp HPH, Pettersen A, Hauge A, Breedveld GD (2008b) Assessing PAH and PCB emissions from the relocation of harbour sediments using equilibrium passive samplers. Chemosphere 72:1581–1587
Davison W, Zhang H (1994) In situ speciation measurements of trace components in natural waters using thin-film gels. Nature 367:546–548
Du Laing G, Rinklebe J, Vandecasteele B, Meers E, Tack FMG (2009) Trace metal behaviour in estuarine and riverine floodplain soils and sediments: a review. Sci Total Environ 407:3972–3985
Eek E, Cornelissen G, Kibsgaard A, Breedveld GD (2008) Diffusion of PAH and PCB from contaminated sediments with and without mineral capping; measurement and modelling. Chemosphere 71:1629–1638
Eek E, Cornelissen G, Breedveld GD (2010) Field measurement of diffusional mass transfer of HOCs at the sediment-water interface. Environ Sci Technol 44:6752–6759
Esteve-Turrillas FA, Yusa V, Pastor A, de la Guardia M (2008) New perspectives in the use of semipermeable membrane devices as passive samplers. Talanta 74:443–457
Förstner U, Salomons W (1988) Dredged materials. In: Salomons W, Bayne BL, Duursma EK, Förstner U (eds) Pollution of the North Sea, Springer–Verlag, Berlin, pp 225–245
Gabriel KR (1971) Biplot graphic display of matrices with application to principal component analysis. Biometrika 58:453–467
Gale R (1998) Three-compartment model for contaminant accumulation by semipermeable membrane devices. Environ Sci Technol 32:2292–2300
Garmo O, Royset O, Steinnes E, Flaten T (2003) Performance study of diffusive gradients in thin films for 55 elements. Anal Chem 75:3573–3580
Gourlay-France C, Lorgeoux C, Tusseau-Vuillemin MH (2008) Polycyclic aromatic hydrocarbon sampling in wastewaters using semipermeable membrane devices: accuracy of time-weighted average concentration estimations of truly dissolved compounds. Chemosphere 73:1194–1200
Harman C, Bøyum O, Tollefsen KE, Thomas KV, Grung M (2008) Uptake of some selected aquatic pollutants in semipermeable membrane devices (SPMDs) and the polar organic chemical integrative sampler (POCIS). J Environ Monit 10:239–247
Harman C, Thomas KV, Tollefsen KE, Meier S, Bøyum O, Grung M (2009) Monitoring the freely dissolved concentrations of polycyclic aromatic hydrocarbons (PAH) and alkylphenols (AP) around a Norwegian oil platform by holistic passive sampling. Mar Pollut Bull 58:1671–1679
Harman C, Brooks S, Sundt RC, Meier S, Grung M (2011) Field comparison of passive sampling and biological approaches for measuring exposure of PAH and alkylphenols from offshore produced water discharges. Mar Pollut Bull 63:141–148
Hawker DW (2010) Modelling the response of passive samplers to varying ambient fluid concentrations of organic contaminants. Environ Toxicol Chem 29:591–596
Hedge LH, Knott NA, Johnston EL (2009) Dredging related metal bioaccumulation in oysters. Mar Pollut Bull 58:832–840
Huckins JN, Tubergen MW, Manuweera GK (1990) Semipermeable-membrane devices containing model lipid—a new approach to monitoring the bioavailability of lipophilic contaminants and estimating their bioconcentration potential. Chemosphere 20:533–552
Huckins JN, Manuweera GK, Petty JD, Mackay D, Lebo JA (1993) Lipid-containing semipermeable-membrane devices for monitoring organic contaminants in water. Environ Sci Technol 27:2489–2496
Huckins JN, Petty JD, Lebo JA, Almeida FV, Booij K, Alvarez DA, Clark RC, Mogensen BB (2002) Development of the permeability/performance reference compound approach for in situ calibration of semipermeable membrane devices. Environ Sci Technol 36:85–91
Huckins JN, Petty JD, Booij K (2006) Monitors of organic chemicals in the environment. Springer, New York, p 223
Lebo JA, Gale RW, Petty JD, Tillitt DE, Huckins JN, Meadows JC, Orazio CE, Echols KR, Schroeder DJ (1995) Use of the semipermeable-membrane device as an in-situ sampler of waterborne bioavailable PCDD and PCDF residues at sub-parts-per-quadrillion concentrations. Environ Sci Technol 29:2886–2892
Lepland AI, Andersen TJ, Lepland AA, Arp HPH, Alve E, Breedveld GD, Rindby A (2010) Sedimentation and chronology of heavy metal pollution in Oslo harbour, Norway. Mar Pollut Bull 60:1512–1522
Løken AM, Nøland SA, Tidemand N (2007) Uavhengig revisjon av Secora AS og Oslo HAV prosjekt. Veritas Report 2007–1626, p 50
Magnusson J, Andersen T, Amundsen R, Berge JA, Beylich B, Bjerkeng B, Bjerknes E, Gjøsæter J, Grung M, Holth TF, Hylland K, Johnsen T, Lømsland ER, Paulsen Ø, Rønning I, Sørensen K, Schøyen M, Walday M (2008) Monitoring the pollution status of the inner Oslofjord 2007. Norwegian Institute for Water Research, Report 5637–2008. ISBN: 978-82-577 5372–6, p 116 (www.niva.no)
Martino M, Turner A, Nimmo M, Millward GE (2002) Resuspension, reactivity and recycling of trace metals in the Mersey Estuary, UK. Mar Chem 77:171–186
Millward GE, Liu YP (2003) Modelling metal desorption kinetics in estuaries. Sci Total Environ 314–316:613–623
Næs K, Oug E (1997) Multivariate approach to distribution patterns and fate of polycyclic aromatic hydrocarbons in sediments from smelter-affected Norwegian fjords and coastal waters. Environ Sci Technol 31:1253–1258
Oen A, Hauge A (2007) Overvåking av forurensing ved mudring og deponering. Miljøregnskap for nedføring i dypvannsdeponiet i perioden januar til juni 2007. Norwegian Geotechnical Institute (NGI), Oslo, Norway. Report 20051785–31, p 17
Pettersen A, Breedveld G (2009) Dypvannsdeponi ved malmøykalven. Sluttrapport del 1: Miljøkvalitet. Norwegian Geotechnical Institute, Oslo, Norway. Report 20051785–65. p 69 (http://www.renoslofjord.no)
Ranneklev S, Allan I, Enge E (2009) Kartlegging av miljøgifter i Alna og Akerselva. SFT, Oslo, Norway. Report OR-5776, ISBN 978-82-577-5511-9, p 116 (www.niva.no)
Schaanning MT, Bjerkeng B (2001) Remediation of contaminated sediments in Oslo Harbour. Modelling mobilisation of contaminants during deposition in deep anoxic basin. Norwegian Institute for Water Research. Report 4438, ISBN 82-577-4082-9, p 49 (www.niva.no)
Schaanning M, Helland A, Lindholm O, Nilsson HC, Vogelsang C (2005) Mass balance budgets for pollutants in sediment remediation areas in Oslo Harbour. Report 5154, ISBN 82-577-4868-4, p 39 (www.niva.no)
Schaanning M, Breyholz B, Skei J (2006a) Experimental results on effects of capping on fluxes of persistent organic pollutants (POPs) from historically contaminated sediments. Mar Chem 102:46–59
Schaanning M, Bjerkeng B, Helland A, Høkedal J (2006b) Subsea sediment disposal at Malmøykalven, Norway. Spreading of particles and associated contaminants during pilot test dumping. Norwegian Institute for Water Research. Report 5221, ISBN 82-577-4942-7, p 44 (www.niva.no)
SFT (2007) Guidelines on classification of environmental quality in fjords and coastal waters. Norwegian Pollution Control Authority, Oslo, Norway. TA-2229/2007, ISBN 978-82-7655-537-0, p 12
Skei J, Nilsson HC (2008) Chemical analyses of sediment cores from the disposal site Malmøykalven and surrounding areas. Norwegian Institute for Water Research. Report 5614, ISBN 978-82-577-5349-8, p 56 (www.niva.no)
Skei J, Magnusson J, Eek E, Eggen A, Hauge A (1999) Current measurements and sediment investigations in an anoxic basin in inner Oslofjord-Malmøykalven. Norwegian Institute for Water Research, Oslo, Norway, Report 4019–99, ISBN 82-577-3615-5, p 25 (www.niva.no)
Thibodeaux LJ, Huls H, Ravikrishna R, Valsaraj KT, Costello M, Reible DD (2004) Laboratory simulation of chemical evaporation from dredge-produced sediment slurries. Environ Eng Sci 21:730–740
Turner A, Nimmo M, Thuresson KA (1998) Speciation and sorptive behaviour of nickel in an organic-rich estuary (Beaulieu, UK). Mar Chem 63:105–118
Warnken K, Zhang H, Davison W (2007) In situ monitoring and dynamic speciation measurements in solution using DGT. In: Greenwood R, Mills G, Vrana B (eds) Passive sampling techniques in environmental monitoring. Compr Anal Chem 48:251–278
Wenning RJ, Sorensen M, Magar VS (2006) Importance of implementation and residual risk analyses in sediment remediation. Integr Environ Assess Manag 2:59–65
Zhang H, Davison W (1995) Performance-characteristics of diffusion gradients in thin-films for the in-situ measurement of trace-metals in aqueous-solution. Anal Chem 67:3391–3400
Zitko V (1994) Principal component analyses in the evaluation of environmental data. Mar Pollut Bull 28:718–722
Acknowledgements
This study was funded by the Norwegian Climate and Pollution Agency (KLIF), formerly the Pollution Control Authority (SFT). We acknowledge the skills of the crew of RV Trygve Braarud and Aud Helland in the deployment and retrieval of the PSDs and the staff at NIVAs laboratory for performance of all chemical analyses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Gijs D. Breedveld
Rights and permissions
About this article
Cite this article
Schaanning, M.T., Harman, C. & Staalstrøm, A. Release of dissolved trace metals and organic contaminants during deep water disposal of contaminated sediments from Oslo harbour, Norway. J Soils Sediments 11, 1477–1489 (2011). https://doi.org/10.1007/s11368-011-0436-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-011-0436-2