
Abstract
Low mitochondriogenesis is critical to explain loss of muscle function in aging and in the development of frailty. The aim of this work was to explain the mechanism by which mitochondriogenesis is decreased in aging and to determine to which extent it may be prevented by exercise training. We used aged rats and compared them with peroxisome proliferator-activated receptor-γ coactivator-1α deleted mice (PGC-1α KO). PGC-1α KO mice showed a significant decrease in the mitochondriogenic pathway in muscle. In aged rats, we found a loss of exercise-induced expression of PGC-1α, nuclear respiratory factor-1 (NRF-1), and of cytochrome C. Thus muscle mitochondriogenesis, which is activated by exercise training in young animals, is not in aged or PGC-1α KO ones. Other stimuli to increase PGC-1α synthesis apart from exercise training, namely cold induction or thyroid hormone treatment, were effective in young rats but not in aged ones. To sum up, the low mitochondrial biogenesis associated with aging may be due to the lack of response of PGC-1α to different stimuli. Aged rats behave as PGC-1α KO mice. Results reported here highlight the role of PGC-1α in the loss of mitochondriogenesis associated with aging and point to this important transcriptional coactivator as a target for pharmacological interventions to prevent age-associated sarcopenia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent advances in medical care as well as in basic gerontology have led to a significant increase in longevity of populations (life span). However, the remarkable increase in life span has also led to an important increase in frailty and dependency (Gill et al. 2002). It is clear now that we must aim at increasing health span. Muscle aging is a key component of the increase in frailty in human and animal populations (Vanitallie 2003). Physical exercise is an obvious anti-aging mechanism and it is intended to serve as a prevention of cardiovascular aging but also as a prevention of sarcopenia as well as loss in muscle functionality (Fiatarone et al. 1994).
Early work by Miquel et al. proposed that loss of mitochondriogenesis was critical in the fundamental process of aging (Miquel et al. 1980; Miquel 1992). Later, in the 1990s, we reported that mitochondrial damage is an early event in cellular aging (Sastre et al. 1996). This was independently confirmed by the group of Bruce Ames (Hagen et al. 1997). In 2003, it was shown in skeletal muscle that age causes a decrease in ATP content and production by approximately 50% in isolated rat mitochondria (Drew et al. 2003). Since the promotion of mitochondriogenesis is critical to prevent aging, an obvious approach was to try and enhance it by physical exercise (Holloszy and Booth 1976; Davies et al. 1982). Researches have identified the peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) as the master regulator of mitochondriogenesis in mammalian tissues (Puigserver and Spiegelman 2003; Wu et al. 1999; Puigserver et al. 1998). In vivo and in vitro studies have shown that PGC-1α levels stimulate mitochondrial proliferation in skeletal muscle (Hood et al. 2006). Increased PGC-1α levels in skeletal muscle by using transgenic MCK-PGC-1α mice (PGC-1α driven by a muscle creatine kinase promoter), prevents muscle wasting by reducing apoptosis, autophagy, and proteasome degradation (Wenz et al. 2009). Moreover, in a recent study by Henriette Pilegaard’s group, it has been shown that PGC-1α is required for the beneficial effects of moderate exercise training at advanced age to maintain mitochondrial metabolic and antioxidant capacity (Leick et al. 2010). These studies suggest that the modulation of PGC-1α levels in skeletal muscle present an avenue for the prevention and treatment of age-related disorders.
The aim of our work was to explain the mechanism by which mitochondriogenesis is decreased in aging and to determine to which extent it may be prevented by exercise training. As endurance training is known to upregulate PGC-1α expression in young skeletal muscle (Gomez-Cabrera et al. 2008a), modulation of PGC-1α levels by endurance training in aged skeletal muscle may be a very effective strategy for the prevention and treatment of sarcopenia. For our purpose, we used aged rats and compared them with PGC-1α knockout (KO) mice.
Our results show that muscle from old rats present a marked loss in mitochondriogenesis and that this may be due to a lack of induction of PGC-1α (Puigserver et al. 1998). We find a striking similarity between the response to exercise training in PGC-1α KO mice and in old rats. In young rats, PGC-1α is activated in skeletal muscle not only by training but also by cold exposure or triiodothyronine (T3). We report here that there is an age-associated lack of expression of PGC-1α in response to exercise training or to any of the other stimuli tested in rat skeletal muscle.
Material and methods
Rats
For the exercise training experiments, 24 male Wistar rats were randomly divided into four experimental groups: young untrained (n = 6), young trained (n = 6), aged untrained (n = 6), and aged trained (n = 6). For the cold induction experiments, 16 male Wistar rats were randomly divided into four experimental groups: young control (n = 4), young exposed to cold (n = 4), aged control (n = 4), and aged exposed to cold (n = 4). For the thyroid hormone experiments, 16 male Wistar rats were randomly divided into four experimental groups: young control (n = 4), young treated with T3 (n = 4), aged control (n = 4), and aged treated with T3 (n = 4). In all the experimental models, the aged animals were 24 months old and the young ones were 3 months old. We chose 24-month-old rats because previous studies have reported that sarcopenia is evident at this age in this species (Hopp 1993).
Mice
The generation and phenotype of PGC-1α KO mice have been described previously (Lin et al. 2004). The genotype of the offspring was assessed by determining the presence of either a wild type (WT)- or KO-specific DNA fragment after extraction of DNA from a tail piece by the phenol-chloroform/isoamyl method, amplification of fragments by PCR using specific primers and separation on an agarose gel. Analysis of the PGC-1α expression revealed that its mRNA was absent in the skeletal muscle of the PGC-1α KO mice. We also wanted to check the PGC-1α protein levels in the skeletal muscle of the KO mice. For this purpose and in order to prevent unspecific cross-reactivity of PGC-1α antibody in the PGC-1α KO mice, we immunoprecipitated the samples of the KO and WT animals (Fig. 1a). Immunoprecipitation was performed with Dynabeads protein A (Invitrogen) according to the manufacturer’s instructions. The incubation of the antibody (anti-PGC-1, Cayman) with the beads and the incubation of the extract with antibody cross-linked to the beads were both carried out overnight. The PGC-1 IP fractions were then analyzed by Western Blotting. The band of PGC-1α (~92 KDa) was clearly present in the skeletal muscle of the wild-type animals and was absent in the skeletal muscle of the PGC-1α KO mice. Although a faint band was present with a molecular weight over 100 KDa in the PGC-1α KO muscles, taking into account its molecular weight, we do not consider that this band represents PGC-1α. PGC-1β is a very close homolog of PGC-1α and shares extensive sequence identity (Lin et al. 2002). In addition to their similarities, PGC-1α and PGC-1β share common protein binding partners and the regulation of certain gene programs in skeletal muscle (Handschin et al. 2007). This is why we consider that this band could be PGC-1β. However and to further test the effect of PGC-1α deletion on muscle structure, we performed an electron microscopy analysis of soleus muscle from WT and PGC-1α KO animals. Soleus muscle was dissected and fixed overnight in 2% glutaraldehyde, 1% paraformaldehyde, and 0.08% sodium cacodylate buffer. The tissues were post-fixed in 1% osmium tetroxide, dehydrated in graded ethanol, embedded in Poly Bed plastic resin, and sectioned for electron microscopy. Electron microscopic analysis revealed fewer and smaller mitochondria in soleus muscle of PGC-1α KO mice compared to sex- and age-matched WT ones (see Fig. 2).

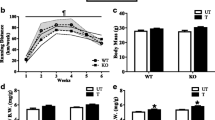
Exercise-induced activation of the mitochondrial biogenesis pathway in skeletal muscle. Western blotting analysis to detect PGC-1α (a and d), NRF-1 (b and e), and cytochrome C (c and f) in PGC-1α KO mice and aged rats. Twenty-seven male mice were randomly divided into four experimental groups: WT untrained (n = 6), WT trained (n = 8), KO untrained (n = 7), and KO trained (n = 6). Twenty-four male Wistar rats were randomly divided into four experimental groups: young untrained (n = 6), young trained (n = 6), aged untrained (n = 6), and aged trained (n = 6). Representative blots are shown. For the densitometric analysis of the results, values are shown as mean (±SD).The content of α-actin, a housekeeping protein marker in skeletal muscle, was determined in all the experimental groups

Electron microscopy analysis of soleus muscle from WT and PGC-1α KO animals. Smaller mitochondria were found in the muscle of PGC-1α KO mice compared to sex- and age-matched WT
Twenty-seven male mice (14 wild-type and 13 PGC-1α KO) were randomly divided into four experimental groups: WT untrained (n = 6), WT trained (n = 8), KO untrained (n = 7), and KO trained (n = 6). The animals were kindly provided from the Centro Nacional de Investigaciones Cardiovasculares Carlos III (Madrid. Spain). The animals were 5–6 months old at the beginning of the experimental protocol.
All animals were fed an ad libitum laboratory diet (Global diet 2,014 l; Harlan Teklad, Madison, WI, USA) and were maintained at 23°C under a light/dark cycle of 12/12 h. The experimental protocol was approved by the Committee on Ethics in Research of the Faculty of Medicine of the University of Valencia, Spain.
Training protocols
Endurance-trained young and aged rats were exercised 5 day/week on an animal treadmill (Model 1050 LS Exer3/6; Columbus Instruments, Columbus, OH, USA) at a relative intensity of 75% VO2max. The treadmill grade and velocity for each experimental group were chosen based on previous studies performed in young and aged rats (Powers et al. 1994; Lawler et al. 1993; Patch and Brooks 1980). During all the experiments, the grade of the treadmill corresponded to 15% for young rats and 5% for aged rats. We followed a modification of the protocol of Davies et al. (1981). The young animals were required to run, the first training session, for 25 min at a speed of 26.8 m × min−1. The old animals were required to run, the first training session, at a speed of 15 m × min−1 for 15 min. The duration an intensity of each work period was increased progressively. The last day of the training week 3, young animals were running for 1 h at a speed of 30 m × min−1 and the aged ones were running for 45 min at a speed of 18 m × min−1. It has been shown that young untrained rats require approximately 75% of their VO2max to run at 26.8 m × min−1 (15% grade) on a treadmill (Patch and Brooks 1980) (Davies et al. 1981). Aged untrained animals require approximately 75% of their VO2max to run at 15 m × min−1 (5% grade) on a treadmill (Lawler et al. 1993). Exercise motivation was provided for all rodents by means of an electronic shock grid at the treadmill rear. However, the electric shock was used sparingly during training and during the endurance capacity test. The untrained groups were exercised at the same speeds for only 10 min every 3 days for the entire 3-week period. Endurance capacity was assessed, before and after the training period, during a run to exhaustion at 26.8 m × min−1 at a grade of 15% for young rats and at 15 m × min−1 at a grade of 5% for aged rats (Davies et al. 1982; see Table 1a).
PGC-1α KO and WT male mice were randomly allocated to either a training group or a control group. The training groups completed 4 weeks of treadmill exercise training five times per week and progressively increased until 60 min at 20 m × min−1 (10% slope) at the end of the second week. Endurance capacity was assessed, before and after the training period, during a run to exhaustion at 20 m × min−1 at a grade of 10% (sees Table 1b). After the tests, the animals were given 48 h of complete rest before being sacrificed.
Cold exposure protocol
After an acclimatization period (1 week), young and aged Wistar male rats were randomly divided into two groups: cold-exposed animals (4 ± 1°C for 24 h) and control animals (24 ± 1°C; Puigserver et al. 1998). Skeletal muscles were removed immediately after the end of the cold exposure.
T3 treatment
Young and aged Wistar male rats were injected intraperitoneally one dose with either T3 (0.4 mg × kg−1) or vehicle (0.9% NaCl-propylene glycol; 40:60 vol/vol). Skeletal muscles were removed 6 h after the injections (Irrcher et al. 2003). Gastrocnemius and soleus muscles were removed quickly, freeze-clamped immediately, and stored at −80°C. All the animals were sacrificed by an overdose of sodium pentobarbital.
SDS-PAGE and Western Blotting
Aliquots of muscle lysates (Ji et al. 2004) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The whole gastrocnemius was used to ensure homogeneity. Proteins were then transferred to nitrocellulose membranes, which were incubated overnight at 4°C with appropriate primary antibodies: anti-PGC-1 (1:1,000, Cayman), anti-NRF-1 (1:200, Santa Cruz Biotechnology Inc.), anti-cytochrome C (1:1,000, Santa Cruz Biotechnology Inc.) and anti-α-actin (1:700, Sigma Aldrich). Thereafter, membranes were incubated with a secondary antibody for 1 h at room temperature. Specific proteins were visualized by using the enhanced chemiluminescence procedure as specified by the manufacturer (Amersham Biosciences, Piscataway, NJ, USA). Autoradiographic signals were assessed by using a scanning densitometer (BioRad, Hercules, CA, USA).
Protein carbonylation
Oxidative modification of total proteins was assessed by immunoblot detection of protein carbonyl groups using the “OxyBlot” protein oxidation kit (Intergen) as previously described (Romagnoli et al. 2010).
Statistics
Results are expressed as mean ± SD. Normality of distribution was checked with the Kolmogorov test and homogeneity of variance was tested by Levene’s statistics. For endurance capacity, a repeated measures two-factor analysis of variance was performed. Repeated measures were performed for training (before training compared with after training); the second factor was the status of animals (control or trained). The main effect of training was tested with Newmann–Keuls post hoc test. For Western Blot analysis, we used a two-way analysis of variance and Bonferroni’s post hoc test (Sigma Stats, version 3.11). Results were considered statistically significant for p < 0.05.
Results
Effect of aging or PGC-1α deletion on endurance response to training
Running time to exhaustion during an endurance capacity test was approximately 63% lower in aged than in young rats (Table 1a). Similarly, running time to exhaustion was ~65–70% lower in the PGC-1α KO mice than in WT animals (Table 1b). These results are in accordance with previous studies demonstrating that endurance capacity was lower in PGC-1α KO animals than in WT (Leick et al. 2008). Table 1a also shows that the intensity and duration of the training regimen followed by young and aged rats was enough to induce a significant improvement in maximal endurance capacity (~200% and ~135%, respectively). Similarly, the training protocol induced an increase in the endurance capacity both in WT and PGC-1α KO mice (~284% and ~173%, respectively; Table 1b). It cannot be ruled out that the differences found on the endurance capacity test, between the young and old rats, may be explained by the different durations of the exercise training protocols, although this is unlikely. Both groups improved their endurance capacity following a similar exercise intensity training protocol (~75% of their VO2max). The different durations of the training periods might explain, in part, some of our results (see “Material and methods” section).
Muscle mitochondriogenesis in PGC-1α KO and aged animals
Figure 1 shows that muscle mitochondriogenesis is considerably impaired in PGC-1α-deficient mice. Figure 1a show that training caused an increase in PGC-1α content in WT mice. As expected, we did not find any PGC-1α protein levels in sedentary or in trained PGC-1α KO animals. Although the band corresponding to the PGC-1α protein was absent in our KO mice (92 kDa), a faint band, which is likely to be PGC-1β, appeared in the Western Blotting over 100 kDa (see “Material and methods” section).
Figure 1b shows that training increased nuclear respiratory factor-1 (NRF-1), a critical intermediate of the mitochondriogenic pathway in WT animals but not in PGC-1α KO ones. It is also shown that sedentary PGC-1α KO animals have considerably less NRF-1 than controls. Mitochondrial content can be measured directly, using morphometric estimates of organelle volume in relation to total cellular volume. More commonly, it is estimated by the change in maximal activity, measured under optimal conditions in vitro, of a typical “marker enzyme” such as citrate synthase, or by the change in content of a single protein-like cytochrome C (Hood 2001; Terjung 1979). Several authors have used cytochrome C content as a marker of mitochondrial mass (Hood et al. 2006; Leick et al. 2008, 2010) and this is the methodology that we have followed in the present work. Training caused an increase in cytochrome C content in WT but not in PGC-1α KO mice. Cytochrome C content in PGC-1α KO animals was also lower than in WT (Fig. 1c). To sum up, deficiency in PGC-1α resulted in a hampered mitochondriogenic responsiveness to exercise training. In aged rats, we found very similar results, i.e., a loss of exercise-induced increase in the protein levels of PGC-1α, NRF-1 and cytochrome C (Fig. 1d–f). The main idea reported here is that old age resembles PGC-1α deficiency in terms of lack of responsiveness to training. Indeed, young animals showed an increase in the protein levels of PGC-1α after training which did not occur in aged ones (Fig. 1d). In Fig. 1e, we show that NRF-1 content was increased in muscle of young rats after training. This effect was lost when we studied aged animals. Finally, Fig. 1f shows that training increased cytochrome C content in muscle of young rats but again this effect was also lost in aged rats.
Oxidative stress and training in aged animals and PGC-1α KO
As apparent in Fig. 1, many of the adaptations of the skeletal muscle to exercise training in aging are similar to those found in PGC-1α KO animals. Thus, we measured the effect of exercise training on skeletal muscle protein oxidation status in young and aged animals and in WT and PGC-1α KO animals. In Fig. 3, we show protein oxidation in PGC-1α KO (Fig. 3a) and in aged animals (Fig. 3b). PGC-1α KO animals show an increase in resting protein oxidation (Fig. 3a). The same happens in aged animals (Fig. 3b). No effects of training were found in any experimental group.

Muscle protein oxidation in untrained and trained animals. Western blotting analysis to detect carbonylated proteins (MW: 37 kDa). Representative experiments are shown. For the densitometric analysis of the results values are shown as mean (±SD) of a WT untrained (n = 6), WT trained (n = 8), PGC-1α KO untrained (n = 7), and PGC-1α KO-trained animals (n = 6); b young untrained (n = 6), young trained (n = 6), aged untrained (n = 6), and aged-trained animals (n = 6)
Lack of activation of PGC-1α by cold exposure or by thyroid hormone treatment in the aged animal
The experiments reported above showing that the muscle of aged animals did not upregulate the expression of PGC-1α in response to exercise training, led us to think that aging could result in a lack of responsiveness of PGC-1α to other physiological stimuli. Two of the key stimulators of PGC-1α are thyroid hormones (Irrcher et al. 2003) and cold exposure (Puigserver et al. 1998). Figure 4a shows that young animals, when exposed to cold, upregulated the expression of PGC-1α threefold but aged animals did not. The same happens with triiodothyronine (T3; Fig. 4b).

PGC-1α protein levels in skeletal muscle of animals exposed to cold (4 ± 1°C) or treated with triiodothyronine (0.4 mg × kg−1). Representative experiments are shown. For the densitometric analysis of the results values are shown as mean (±SD) of a young control (n = 4), young exposed to cold (n = 4), aged control (n = 4), and aged-exposed to cold (n = 4); b young control (n = 4), young treated with T3 (n = 4), aged control (n = 4), aged treated with T3 (n = 4). The content of α-actin, a housekeeping protein marker in skeletal muscle, was determined in all the experimental groups
Discussion
The major idea in this paper is that aging causes a lack of response of PGC-1α to various stimuli, the most important being exercise training (see Fig. 1), but also to cold exposure or thyroid hormone treatment (see Fig. 4).
The role of mitochondria as key generator of oxidative stress and also target of the damage associated with reactive oxygen species (ROS) production was postulated by Miquel in the 1970s (Johnson et al. 1975). Independent work from our laboratory (Sastre et al. 1996) and that from Bruce Ames’ (Hagen et al. 1997) using both metabolic and flow cytometric approaches provided such evidence. We showed that mitochondria are damaged within cells instead of being frail and damaged during the isolation procedure. A functional muscle that has not lost the capacity to synthesize healthy mitochondria is an important contributor to the prevention of frailty, a major problem in medicine, particularly in geriatrics (Fiatarone et al. 1994; Gill et al. 2010). Thus, understanding the molecular mechanisms of mitochondriogenesis in aging has both theoretical and practical importance. It has been reported that the mitochondrial function is adapted in response to calorie restriction and this adaptation is critically involved in lifespan extension (Anderson et al. 2008). Calorie restriction has been shown to activate PGC-1α (Nisoli et al. 2005; Anderson et al. 2008; Anderson and Weindruch 2009) and it may be an effective strategy in delaying aging-induced cellular phenotypes in skeletal muscle (McKiernan et al. 2010). PGC-1α is critical for the adaptation of muscle mitochondriogenesis to exercise which activates the expression of NRF-1 which in turn, activates TFAM, a factor required for the duplication of mitochondrial DNA (Hood 2001). This led us to think that mitochondriogenesis might be impaired in aging because of the impaired response of PGC-1α in old animals when compared with young ones. To understand the role of the redox-sensitive PGC-1α in the regulation of mitochondriogenesis in muscle, we used animals which were KO for PGC-1α.
We found a striking similarity in the molecular response of exercise in the mitochondriogenic pathway in mice which are deleted of PGC-1α and in the old rats. The whole pathway involving PGC-1α→NRF-1 and finally cytochrome C (an indicator of mitochondrial mass) responded positively to exercise training in young rats, but failed to do so in old ones. This was precisely the same behavior as that in PGC-1α-deleted mice.
It could be argued that the intensity of the exercise training was not enough to onset the mitochondrial biogenesis in PGC-1α KO mice or in old rats. However, Table 1 shows that the intensity and duration of the training regimen followed by our animals was enough to induce a significant improvement in maximal endurance capacity.
PGC-1α protects against skeletal muscle atrophy (Sandri et al. 2006) and very recently it has been shown to be required for training-induced prevention of age associated decline in mitochondria (Leick et al. 2010). Moreover, relevance of PGC-1α in sarcopenia and metabolic diseases during aging has been also suggested (Wenz et al. 2009). Transgenic MCK-PGC-1α animals have preserved mitochondrial function, neuromuscular junctions, and muscle integrity during aging. Moreover, increased PGC-1α levels in skeletal muscle prevent muscle wasting by reducing apoptosis, autophagy, and proteasome degradation (Wenz et al. 2009).
Our studies in exercise adaptations to aging, as stated above, led us to the conclusion that PGC-1α was not responding to exercise training in old animals. We suspected that PGC-1α might not be induced by any kind of stimulus in old animals. To test this hypothesis, we used two well-known stimuli of PGC-1α in young and old rats, namely thyroid hormone stimulation and cold acclimation (Irrcher et al. 2003; Puigserver et al. 1998). Figure 4 shows that both T3 treatment or cold acclimatization caused a very pronounced activation of PGC-1α in young animals. However, there was a striking lack of activation of PGC-1α by any of the stimuli tested when we were using old animals.
It has been reported that the health benefits of chronic exercise may be, at least partially, due to a reduction in mitochondrial oxidant production (Judge et al. 2005). These data question the very well-established idea that exercise generates free radicals. This was first established by the group of Packer (Davies et al. 1982) who showed that ROS are generated during muscle contraction. In that seminal paper, the authors already postulated that the exercise-induced mitochondriogenesis might be stimulated by ROS (Davies et al. 1982). We provided the first clear-cut evidence that exercise generates oxidative stress only when it is exhaustive (Sastre et al. 1992; Gomez-Cabrera et al. 2003). However, physical exercise can be considered as a double edge sword: when practiced strenuously it causes oxidative stress and cell damage but when practiced with moderation, it increases the expression of antioxidant enzymes and thus should be considered as an antioxidant (Gomez-Cabrera et al. 2008b). Here, we have measured the effect of exercise training on skeletal muscle protein oxidation status in young and aged rats and in WT and PGC-1α KO mice. We have found that skeletal muscle from aged and KO PGC-1α animals exhibit oxidative stress, i.e., an increase in protein carbonylation, in resting conditions. The protein oxidation was not significantly increased after training in any experimental group which is in accordance with our idea that exercise training does not increase oxidative stress (Gomez-Cabrera et al. 2008b). Previous work from our laboratory as well as from others has demonstrated that interfering with free radical production with antioxidants may hamper mitochondriogenic activation by exercise (Gomez-Cabrera et al. 2008a; Strobel et al. 2010; Ristow et al. 2009). In the case of aging, we outline a different scenario. Our data show that there are chronic high levels of ROS in the skeletal muscle of old and PGC-1α KO animals. We consider that under these circumstances, the response to exercise of the redox-sensitive cell signaling pathways may be hampered. However, this hypothesis needs to be confirmed in future investigations.
PGC-1α is currently identified as a new therapeutic target for treatment of age-related mitochondrial dysfunction in skeletal muscle, and more generally for sarcopenia (Sandri et al. 2006). Moreover, recently, an interesting paper has underpinned the critical role of PGC-1α to link nuclear and mitcochondrial changes in aging (Kelly 2011). Our study highlights the importance of maintaining a normal PGC-1α responsiveness (which we show here is lost in aging) to maintain normal muscle function, and certainly this problem deserves more research. A schematic interpretation of our results is in Fig. 5.

PGC-1α is not functional in the aged skeletal muscle and it can be involved in the decrease in mitochondrial biogenesis during aging. Proposed mechanism
References
Anderson RM, Weindruch R (2009) Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol Metab 21(3):134–141
Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R (2008) Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 7(1):101–111
Davies KJ, Packer L, Brooks GA (1981) Biochemical adaptation of mitochondria, muscle, and whole-animal respiration to endurance training. Arch Biochem Biophys 209(2):539–554
Davies KJ, Quintanilha AT, Brooks GA, Packer L (1982) Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun 107(4):1198–1205
Drew B, Phaneuf S, Dirks A, Selman C, Gredilla R, Lezza A, Barja G, Leeuwenburgh C (2003) Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol 284(2):R474–R480
Fiatarone MA, O’Neill EF, Ryan ND, Clements KM, Solares GR, Nelson ME, Roberts SB, Kehayias JJ, Lipsitz LA, Evans WJ (1994) Exercise training and nutritional supplementation for physical frailty in very elderly people. N Engl J Med 330(25):1769–1775
Gill TM, Baker DI, Gottschalk M, Peduzzi PN, Allore H, Byers A (2002) A program to prevent functional decline in physically frail, elderly persons who live at home. N Engl J Med 347(14):1068–1074
Gill TM, Gahbauer EA, Han L, Allore HG (2010) Trajectories of disability in the last year of life. N Engl J Med 362(13):1173–1180
Gomez-Cabrera MC, Pallardo FV, Sastre J, Vina J, Garcia-del-Moral L (2003) Allopurinol and markers of muscle damage among participants in the Tour de France. Jama 289(19):2503–2504
Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J, Vina J (2008a) Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr 87(1):142–149
Gomez-Cabrera MC, Domenech E, Vina J (2008b) Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med 44(2):126–131
Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN (1997) Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci USA 94(7):3064–3069
Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM (2007) Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 282(41):30014–30021
Holloszy JO, Booth FW (1976) Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol 38:273–291
Hood DA (2001) Invited review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol 90(3):1137–1157
Hood DA, Irrcher I, Ljubicic V, Joseph AM (2006) Coordination of metabolic plasticity in skeletal muscle. J Exp Biol 209(Pt 12):2265–2275
Hopp JF (1993) Effects of age and resistance training on skeletal muscle: a review. Phys Ther 73(6):361–373
Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA (2003) PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol 284(6):C1669–C1677
Ji LL, Gomez-Cabrera MC, Steinhafel N, Vina J (2004) Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J 18(13):1499–1506
Johnson JE, Mehler WR, Miquel J (1975) A fine structural study of degenerative changes in the dorsal column nuclei of aging mice. Lack of protection by vitamin E. J Gerontol 30(4):395–411
Judge S, Jang YM, Smith A, Selman C, Phillips T, Speakman JR, Hagen T, Leeuwenburgh C (2005) Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am J Physiol Regul Integr Comp Physiol 289(6):R1564–R1572
Kelly DP (2011) Cell biology: ageing theories unified. Nature 470:342–343
Lawler JM, Powers SK, Visser T, Van Dijk H, Kordus MJ, Ji LL (1993) Acute exercise and skeletal muscle antioxidant and metabolic enzymes: effects of fiber type and age. Am J Physiol 265(6 Pt 2):R1344–R1350
Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J, Pilegaard H (2008) PGC-1alpha is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab 294(2):E463–E474
Leick L, Lyngby SS, Wojtasewski JF, Pilegaard H (2010) PGC-1alpha is required for training-induced prevention of age-associated decline in mitochondrial enzymes in mouse skeletal muscle. Exp Gerontol 45(5):336–342
Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM (2002) Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem 277(3):1645–1648
Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119(1):121–135
McKiernan SH, Colman RJ, Lopez M, Beasley TM, Aiken JM, Anderson RM, Weindruch R (2010) Caloric restriction delays aging-induced cellular phenotypes in rhesus monkey skeletal muscle. Exp Gerontol 46(1):23–29
Miquel J (1992) An update on the mitochondrial-DNA mutation hypothesis of cell aging. Mutat Res 275(3–6):209–216
Miquel J, Economos AC, Fleming J, Johnson JE Jr (1980) Mitochondrial role in cell aging. Exp Gerontol 15(6):575–591
Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO (2005) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310(5746):314–317
Patch LD, Brooks GA (1980) Effects of training on VO2 max and VO2 during two running intensities in rats. Pflugers Arch 386(3):215–219
Powers SK, Criswell D, Lawler J, Ji LL, Martin D, Herb RA, Dudley G (1994) Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am J Physiol 266(2 Pt 2):R375–R380
Puigserver P, Spiegelman BM (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24(1):78–90
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92(6):829–839
Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 106(21):8665–8670
Romagnoli M, Gomez-Cabrera MC, Perrelli MG, Biasi F, Pallardo FV, Sastre J, Poli G, Vina J (2010) Xanthine oxidase-induced oxidative stress causes activation of NF-kappaB and inflammation in the liver of type I diabetic rats. Free Radic Biol Med 49(2):171–177
Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM (2006) PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103(44):16260–16265
Sastre J, Asensi M, Gasco E, Pallardo FV, Ferrero JA, Furukawa T, Vina J (1992) Exhaustive physical exercise causes oxidation of glutathione status in blood: prevention by antioxidant administration. Am J Physiol 263(5 Pt 2):R992–R995
Sastre J, Pallardo FV, Pla R, Pellin A, Juan G, O’Connor JE, Estrela JM, Miquel J, Vina J (1996) Aging of the liver: age-associated mitochondrial damage in intact hepatocytes. Hepatology 24(5):1199–1205
Strobel NA, Peake JM, Matsumoto A, Marsh SA, Coombes JS, Wadley GD (2010) Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Med Sci Sports Exerc. doi:10.1249/MSS.0b013e318203afa3
Terjung RL (1979) The turnover of cytochrome C in different skeletal-muscle fibre types of the rat. Biochem J 178(3):569–574
Vanitallie TB (2003) Frailty in the elderly: contributions of sarcopenia and visceral protein depletion. Metabolism 52(10 Suppl 2):22–26
Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT (2009) Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA 106(48):20405–20410
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98(1):115–124
Acknowledgements
This work was supported by grants SAF2008-00270, SAF2009-08334, and BFU2007-65803/BFI from the Spanish Ministry of Education and Science; PROMETEO/2010/074 from the Consellería de Educación de la Generalitat Valenciana. ISCIII2006-RED13-027 from the “Red Temática de Investigación Cooperativa en Envejecimiento y Fragilidad (RETICEF)”, EU Funded COSTB35 and DPS2008-06968 from Spanish Ministry of Innovation and Science. This study has been cofinanced by FEDER funds from the European Union.
Author information
Authors and Affiliations
Corresponding author
Additional information
Frederic Derbré and Mari Carmen Gomez-Cabrera contributed equally to this work.
About this article
Cite this article
Derbré, F., Gomez-Cabrera, M.C., Nascimento, A.L. et al. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. AGE 34, 669–679 (2012). https://doi.org/10.1007/s11357-011-9264-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-011-9264-y